
Abstract
Next-generation sequencing (NGS) technologies have increasingly played crucial roles in biological and medical research, but are not yet in routine use in veterinary diagnostic laboratories. We developed and applied a procedure for high-throughput RNA sequencing of Porcine reproductive and respiratory syndrome virus (PRRSV) from cell culture–derived isolates and clinical specimens. Ten PRRSV isolates with known sequence information, 2 mixtures each with 2 different PRRSV isolates, and 51 clinical specimens (19 sera, 16 lungs, and 16 oral fluids) with various PCR threshold cycle (Ct) values were subjected to nucleic acid extraction, cDNA library preparation (24-plexed), and sequencing. Whole genome sequences were obtained from 10 reference isolates with expected sequences and from sera with a PRRSV real-time reverse transcription PCR Ct ≤ 23.6, lung tissues with Ct ≤ 21, and oral fluids with Ct ≤ 20.6. For mixtures with PRRSV-1 and -2 isolates (57.8% nucleotide identity), NGS was able to distinguish them as well as obtain their respective genome sequences. For mixtures with 2 PRRSV-2 isolates (92.4% nucleotide identity), sequence reads with nucleotide ambiguity at numerous sites were observed, indicating mixed infection; however, individual virus sequences could only be separated when 1 isolate identity and sequence in the mixture is known. The NGS approach described herein offers the prospect of high-throughput sequencing and could be adapted to routine workflows in veterinary diagnostic laboratories, although further improvement of sequencing outcomes from clinical specimens with higher Ct values remains to be investigated.
Introduction
Porcine reproductive and respiratory syndrome (PRRS), characterized by reproductive failure in breeding females and respiratory distress in pigs of all ages, is an economically devastating disease globally. In a 2013 analysis, its economic impact on the U.S. swine industry was estimated at $664 million annually or $1.8 million per day. 12 A significant amount of time, money, and effort has been dedicated to controlling and/or preventing PRRS, including vaccination, herd management, and biosecurity. Nonetheless, the effectiveness of vaccines has been disappointing given the high rate of genetic and antigenic variability of Porcine reproductive and respiratory syndrome virus (PRRSV), the etiologic agent of PRRS.16,25,35
PRRSV (order Nidovirales, family Arteriviridae, genus Arterivirus) is an enveloped, single-stranded, positive-sense RNA virus. 5 The PRRSV genome is ~15 kb in length and contains at least 10 open reading frames (ORFs) in the order of: 5′ untranslated region (UTR), ORF1a/1b, ORF2a, ORF2b, ORF3, ORF4, ORF5a, ORF5, ORF6, ORF7, and 3′-UTR. 38 Among the structural protein genes (ORF2a–ORF7), the ORF5 encoding the major envelope glycoprotein 5 is most variable 38 and is commonly used for sequencing to study molecular epidemiology and/or genetic relatedness of PRRSV.14,24,34,40 PRRSV sequencing has also often been used in making an intervention decision, including vaccine selection by swine practitioners and seed virus selection by vaccine manufacturers.6,10,18,44 However, ORF5 only covers ~4% of the entire genome or 12% of structural genes, and thus may not be able to provide the breadth of evidence needed for differentiating PRRSV strains. In addition, PRRSV strains are divided into at least 2 distinct genotypes: type 1 (European type) and type 2 (North American type), with extensive genetic variability both within and between these genotypes.1,3,11,30,39,45 Moreover, PRRSV has demonstrated continuous and rapid evolution, generating new variants.4,13,28 A rapid, high-throughput, and accurate whole genome sequencing method for PRRSV possesses enormous potential to provide more comprehensive information to diagnosticians, researchers, and the swine industry.
Great progress has been made in nucleic acid sequencing technologies. 26 The first high-throughput next-generation sequencing (NGS) technology, namely Roche 454 FLX platform, became available in 2005 (http://www.454.com). Subsequently, Illumina released the Genome Analyzer in early 2007 (http://www.illumina.com), and Applied Biosystems distributed the SOLiD System 2.0 platform (http://www.appliedbiosystems.com) in 2008. These novel sequencing techniques, characterized by massive sequence output, low cost per base, and short turnaround time, have significantly changed the way we understand pathogens. Theoretically, the metagenomics-based strategy (i.e., hypothesis-free) is able to detect novel pathogens or variants, and simultaneously identify mixed infection of microorganisms, including bacteria, viruses, and parasites. 33 It can also be used to study the dynamics of pathogens and associated microbiota in animal specimens during the course of infectious diseases.9,42 Despite many advantages, the use of NGS in veterinary testing is in its infancy. We aimed to establish a fast, cost-effective, and high-throughput procedure using NGS technology and to evaluate its application to determine the whole genome sequences of PRRSV strains in cell culture–derived isolates and clinical specimens.
Materials and methods
Reference PRRSV isolates
Six PRRSV-2 (VR-2332, VR-2385, NADC20, MN184, ISU-P [ATCC VR-2402], and SDSU73 [ATCC PTA-6322]), 1 PRRSV-1 (Lelystad strain), and 3 PRRSV-2 vaccine (MLV vaccine, a ATP vaccine, b and F vaccine c ) strains were used in the study. These reference strains were selected because they are commonly used in research studies, and their complete genome sequences have been published,3,27,31,32,43 except for NADC20. PRRSV reference strains were propagated in MARC-145 cells, a clone of the African monkey kidney cell line MA-104, 15 and had infectious titers of 104 to 106 TCID50/mL (50% tissue culture infective dose per milliliter) before being used for sequencing.
Mixtures of PRRSV isolates
PRRSV Lelystad isolate and PRRSV VR-2385 isolate were manually mixed in the volume ratio of 1:1 to create a mixture containing a PRRSV-1 and -2 isolate. Similarly, MLV vaccine a and PRRSV VR-2385 isolates were manually mixed in the volume ratio of 1:1 to create a mixture containing both PRRSV-2 vaccine strain and wild-type isolate.
Clinical specimens
Nineteen sera, 16 lungs, and 16 oral fluid samples were selected from submissions to the Iowa State University Veterinary Diagnostic laboratory (ISU VDL; Ames, Iowa) and used in attempts of whole genome sequencing. The samples tested positive with various threshold cycle (Ct) values by a commercial PRRSV real-time reverse transcription (RT)-PCR. d
Total RNA extraction for NGS and RNA quantification
Aforementioned PRRSV reference isolates, mixtures of PRRSV isolates, and clinical samples were used for NGS attempts. For virus isolates, serum samples, and oral fluids, 300 µL of each sample were centrifuged at 4,200 × g for 30 min at 4°C to remove host cells and/or other debris. Two hundred microliters of supernatants were then taken for total viral RNA extraction. For lung tissues, 10% (wt/vol) homogenates were made in Earle balanced salt solution. e Ten milliliters of each homogenate were then centrifuged at 4,200 × g for 30 min at 4°C. All of the supernatants were further filtered through a 0.22-μm membrane filter and then centrifuged at 180,000 × g at 4°C for 3 h. Next, the supernatants were discarded, and resulting pellets were resuspended in 200 µL of sterile phosphate-buffered saline (PBS; 1× pH 7.4) f for total viral RNA extraction. To extract total RNA from the preprocessed samples, a magnetic particle processor g and a viral RNA isolation kit h were used following the manufacturer’s instructions, with minor modifications. Briefly, 200 μL of each sample and 400 μL of lysis/binding solution without carrier RNA were added into wells along with 20 μL of bead-enhancer mix. After 2 washes with 300 µL of wash solution I and 2 washes with 450 µL of wash solution II, RNAs were eluted in 50 µL of RNase-free water and stored at −80°C until use. Alternatively, total RNA extraction was performed using a column-based total RNA purification kit. i This method produced a similar RNA yield and quality as did the semiautomatic RNA extraction method with the viral RNA isolation kit h described above. A spectrophotometer j and a RNA assay kit k were used to quantify the RNA in extracts.
Library preparation for NGS
Complementary (c)DNA libraries were constructed from total RNA using a total RNA sample preparation kit l following the low-sample protocol in manufacturer’s guidelines, with some modifications. The library preparation is summarized in the following 6 steps.
Step 1: rRNA depletion
For the total RNA from lung tissue, ribosomal (r)RNA depletion was performed. Briefly, 0.1–1 μg of total RNA was diluted with nuclease-free water to a final volume of 10 μL. Ribosomal RNA was removed using a commercial kit. m Finally, 8.5 μL of clean RNA was used in the next steps. Ribosomal RNA depletion was not performed on total RNA extracted from cell culture–derived isolates, serum, or oral fluid samples.
Step 2: RNA fragmentation
The time for fragmentation was optimized to 2 min to generate a median insert size of 340 nucleotides. Distribution of size of RNA fragment was confirmed using a bioanalyzer. n The fragmentation mixture was immediately subjected to cDNA synthesis.
Step 3: cDNA synthesis
The first-strand cDNA was synthesized using random primers (hexamer) and reverse transcriptase. The RNA template was then removed and a replacement strand was synthesized to generate double-stranded cDNA. These procedures were performed according to the low-sample protocol without in-line control reagent followed by DNA cleanup using a commercial kit. o
Step 4: 3′-end adenylation and adapter ligation
A single “A” nucleotide was added to the 3′-end of the blunt fragments, and multiple indexing adapters were ligated to the fragment by the complementary overhang with the corresponding single “T” nucleotide at the 3′-end of the adapter. Up to 24 indexing adapters were used to distinguish 24 different samples. DNA was then cleaned up twice using a commercial kit. o
Step 5: Enrichment of DNA fragments
DNA fragments with adapter molecules at both ends were PCR amplified, and the PCR products were cleaned up using a commercial kit. o PCR conditions were 98°C for 30 s, 15 cycles of 98°C for 10 s, 60°C for 30 s and 72°C for 30 s, and 72°C for 5 min.
Step 6: Library quantification, normalization, pooling, and sequencing
Libraries were analyzed for size distribution using a bioanalyzer n and further quantified by a PRRSV real-time RT-PCR using a quantification kit p in accordance with the manufacturer’s protocol. Multiplex libraries were then normalized to 10 nM with nuclease-free water and pooled in an equal volume before loading to a flow cell. The pooled libraries were sequenced on a NGS sequencer q with 150-bp end reads by following the operation manual at the ISU DNA Facility (Ames, Iowa).
NGS data analysis
Keywords “PRRSV complete genome” were used to search against the NCBI nucleotide database, resulting in 459 PRRSV whole genome sequences that were saved as a reference genome library for reads mapping. The adapter-filtered original paired-end reads were mapped against the PRRSV reference genome library using mapping software.r,20 Sequence IDs of mapped reads were extracted using a sequence alignment tool.s,21 The new paired-end read sets were extracted from the original paired-end FASTQ files using seqtk (https://github.com/lh3/seqtk) with the list of mapped IDs to ensure both reads of 1 pair were selected. Then, the mapped read sets were used for de novo assembly using assembly software.t,37 k-mer sized from 20 to 64 were tested, and the best assembly was chosen according to the maximal contig. If a single contig of full-length genome was not achieved after de novo assembly, all contigs exported from the assembly software t were reassembled with or without a reference genome sequence using another software. u If a consensus sequence of full-length genome could not be assembled without a reference, the reference genome obtained via BLAST search of the longest contig was added to the software u to yield a whole genome consensus. Then, the reference genome sequence was deleted from the assembly, resulting in a consensus genome with gaps properly annotated.
For analyzing sequence data generated from a mixture containing PRRSV-2 MLV vaccine strain and PRRSV-2 VR-2385 isolate, analysis procedures described above (mapping against the PRRSV reference genome library followed by de novo assembly) were first performed. Subsequently, Integrative Genomics Viewer (IGV)36,41 was used to align sequence reads against the obtained consensus sequences. This process enables visualization of nucleotides at each position of sequencing reads.
PRRSV real-time RT-PCR tests and PRRSV ORF5 sequencing
Nucleic acids were extracted from serum (50 µL), lung homogenate (50 µL), and oral fluids (100 µL) using a commercial RNA/DNA extraction kit v and an automatic instrument g following the instructions of the manufacturer. Nucleic acids were eluted into 90 µL of elution buffer. A commercial PRRSV real-time RT-PCR d was used to test samples for the presence or absence of PRRSV RNA according to the manufacturer’s instructions using a real-time PCR instrument. w
PRRSV ORF5 sequencing was performed using different procedures depending on virus genotypes. For PRRSV-2, a primary primer set (forward primer P5F2 5′-AAGGTGGTATTTGGCAATGTGTC-3′ and reverse primer P5R2 5′-GAGGTGATGAATTTCCAGGTTTCTA-3′) was used for RT-PCR amplification of a fragment (1,082 bp) covering PRRSV ORF5 and 5′- and 3′-flanking regions using a 1-step RT-PCR kit x following the cycling conditions of: 48°C for 20 min; 94°C for 3 min; 45 cycles of 94°C for 30 s, 50°C for 50 s, and 68°C for 50 s; and 68°C for 7 min. Detection of PCR products was conducted with an advanced detection system. y If the PCR products with desired size (~1,082 bp) were obtained, the products were purified using a commercial kit z and submitted to the ISU DNA Facility for sequencing. Sequence data were analyzed using software u described above. If products of correct size were not obtained, alternative primers (forward primer ORF5U 5′-GGTGGGCAACKGTTTTAGCCTGTC-3′ and reverse primer ORF5L 5′-GGTAATAGARAAYGCCAAAAGCACC-3′) were used to RT-PCR amplify the ORF5 and 5′-and 3′-flanking regions using a one-step RT-PCR kit x with the same cycling conditions as described for the primary primer set except that the annealing temperature was changed to 52°C. If the products of correct size (723 bp) were obtained, the products were purified and sequenced as described above; if the correct-size products were not obtained, the sample was considered unsuccessful for ORF5 sequencing.
For PRRSV-1, forward primer L1F 5′-TGAGGTGGGCTACAACCATT-3′ and reverse primer L1R 5′-AGGCTAGCACGAGCTTTTGT-3′ and a 1-step RT-PCR kit x were used to amplify the PCR product. The cycling conditions were the same as described for the PRRSV-2 primary primer set except that the annealing temperature was changed to 55°C. If the products with the expected size of 702 bp were obtained, the products were purified and sequenced as described above; if not, the sample was considered unsuccessful for ORF5 sequencing.
Results
Next-generation sequencing outcomes on PRRSV reference isolates
Ten well-characterized PRRSV isolates were chosen as reference strains to optimize the NGS procedures, including 6 PRRSV-2 isolates, 1 PRRSV-1 isolate, and 3 vaccine strains. Full-length genome sequences were obtained from all 10 PRRSV reference isolates using NGS technology (Table 1). The total reads of PRRSV isolates ranged from 91,932 to 1,726,206, and ~1.87–74.2% of the total reads were mapped to a reference PRRSV genome library. Although the percentage of mapped reads was only 1.87% for isolate VR-2385, its number of mapped reads was 31,702, and a full-length genome of 14,956 nucleotides (nt) was still assembled. The smallest number of mapped reads was 6,090 from the NADC20 isolate, representing ~61× coverage of the PRRSV genome. The whole genome sequences of the 10 virus isolates obtained using NGS technology in our study, except NADC20, were compared with the corresponding sequences available in GenBank, which were determined by the traditional Sanger method (Table 1). The complete genome sequence of the F vaccine was not available in GenBank; however, its parental strain PRRSV P129 sequence is available, which was used for comparison to the NGS data. The nucleotide identities between the NGS sequences and the previously reported sequences ranged from 99.3–99.9% for all PRRSV isolates evaluated.
Summary of custom* RNA-Seq next-generation sequencing (NGS) on Porcine reproductive and respiratory syndrome virus (PRRSV) reference isolates.
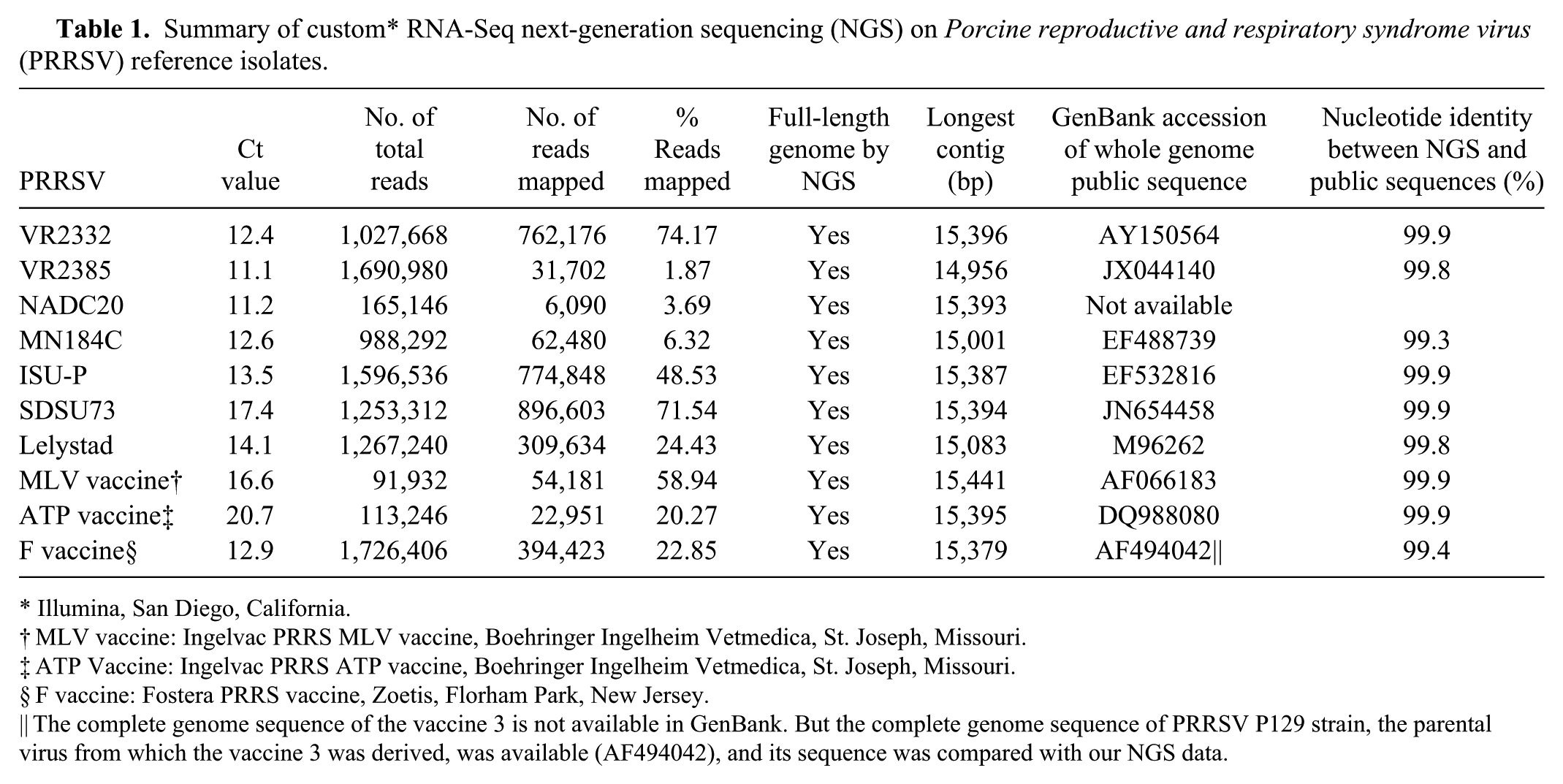
Illumina, San Diego, California.
MLV vaccine: Ingelvac PRRS MLV vaccine, Boehringer Ingelheim Vetmedica, St. Joseph, Missouri.
ATP Vaccine: Ingelvac PRRS ATP vaccine, Boehringer Ingelheim Vetmedica, St. Joseph, Missouri.
F vaccine: Fostera PRRS vaccine, Zoetis, Florham Park, New Jersey.
The complete genome sequence of the vaccine 3 is not available in GenBank. But the complete genome sequence of PRRSV P129 strain, the parental virus from which the vaccine 3 was derived, was available (AF494042), and its sequence was compared with our NGS data.
Next-generation sequencing outcomes on mixtures with 2 PRRSV isolates
For a mixture containing a PRRSV-1 isolate (Lelystad strain) and a PRRSV-2 isolate (VR-2385), 2 sequences corresponding to the Lelystad strain and VR-2385 strain, respectively, were de novo assembled and obtained when raw sequence reads were mapped against the reference PRRSV genome library including 459 PRRSV whole genomes. However, for a mixture containing a PRRSV-2 MLV vaccine strain and a PRRSV-2 wild-type isolate (VR-2385), similar analysis procedures only resulted in 1 consensus full-length genomic sequence; the MLV vaccine and VR-2385 genomic sequences could not be separately assembled. When the IGV software was used to align all sequence reads against the obtained consensus whole genome sequence, nucleotide variations at some positions of raw sequencing reads were observed, indicating a mixed infection. Because nucleotide variation at each single position could be clearly visualized, and identification of the PRRSV strains in the mixture was known, this enabled us to determine which nucleotide corresponded to the MLV vaccine and VR-2385, respectively, at those positions with nucleotide ambiguity.
Next-generation sequencing outcomes on clinical specimens
PRRSV whole genome sequencing was attempted using NGS directly from 19 sera, 16 lungs, and 16 oral fluid samples with different Ct values (Tables 2–4). Ct values of samples had an impact on the outcome and quality of the NGS data. For example, 52.4% (569,911 of 1,087,018) of the total reads was mapped to PRRSV genomes in a serum sample with a Ct value of 15.1, whereas only 0.89% (19,699 of 2,223,592) of the total reads was mapped to PRRSV genomes in a serum sample with a Ct value of 23.6 (Table 2). From serum samples with Ct 15.1–23.6 (n = 10), full-length genomic sequences were successfully obtained without any gaps. From a serum sample with Ct 24.3, most of the genome sequences were obtained except for 8 gaps of <150 bp and 1 gap of <750 bp. From serum samples with Ct 25.0–26.8 (n = 4), some sequence contigs could be assembled, albeit with some gaps at different sizes; full-length sequences were not obtained. When Ct values increased to ≥28, no contigs were obtained for assembly (Table 2).
Summary of custom* RNA-Seq next-generation sequencing outcomes on serum samples.
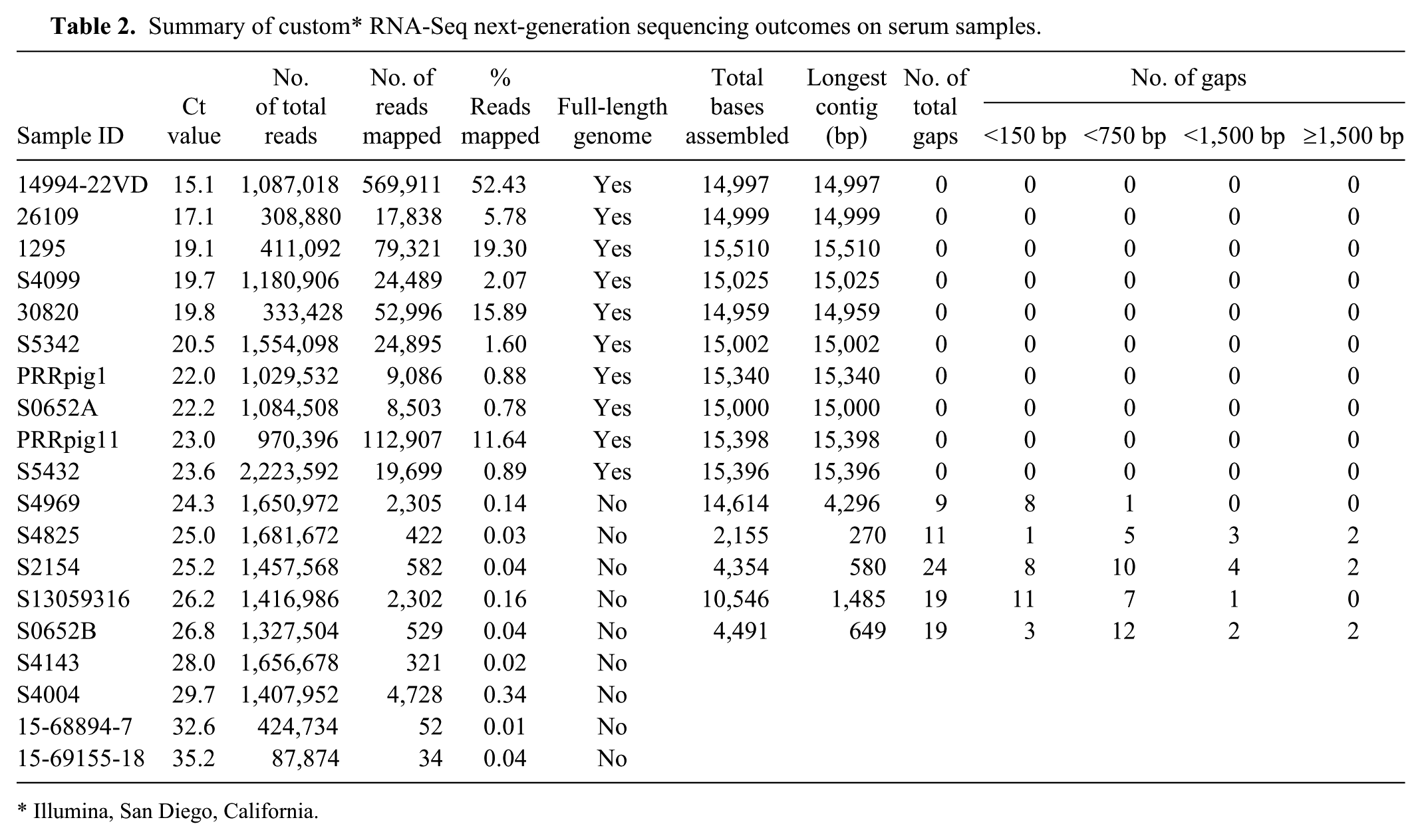
Illumina, San Diego, California.
Summary of custom* RNA-Seq next-generation sequencing outcomes on lung samples.
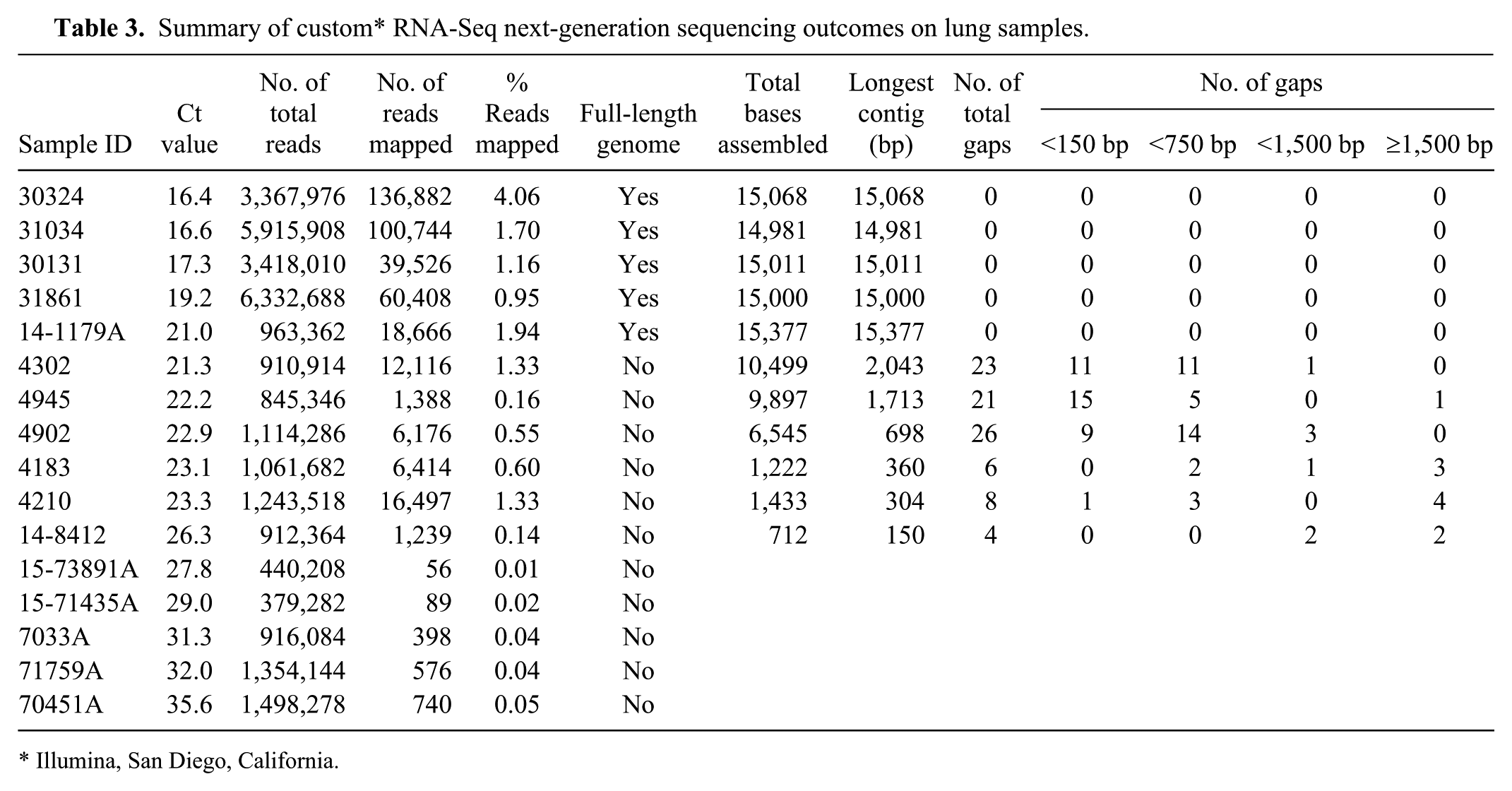
Illumina, San Diego, California.
Summary of custom* RNA-Seq next-generation sequencing outcomes on oral fluid samples.
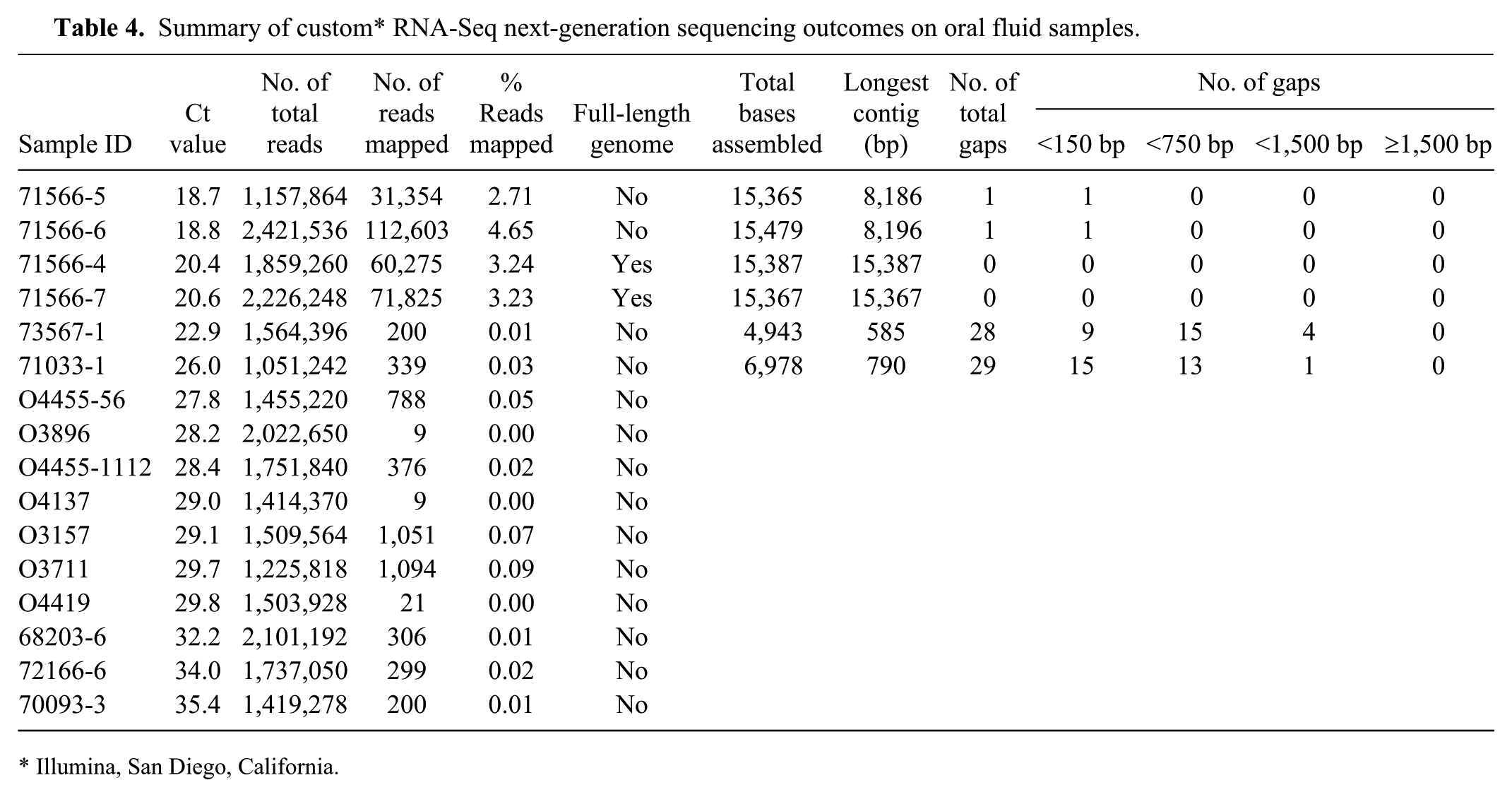
Illumina, San Diego, California.
For lung samples, the total reads that could be mapped to PRRSV reference genomes decreased with increasing real-time RT-PCR Ct values (Table 3). Full-length PRRSV genomic sequences were obtained from lung tissues with Ct values 16.4–21.0 (n = 5). From lung tissues with Ct values of 21.3–26.3 (n = 6), some sequence contigs could be assembled, but gaps were present, and full-length sequences were not obtained. When Ct values increased to ≥27.8, no contigs were obtained for assembly (Table 3).
Whole genome sequencing was attempted on 16 oral fluids with Ct values of 18.7–35.4 (Table 4). From oral fluids with Ct values of 18.7 and 18.8 (n = 2), full-length genomic sequences were obtained, except 1 gap of <150 bp. From oral fluids with Ct values 20.4 and 20.6 (n = 2), full-length genomic sequences without gaps were obtained. From oral fluids with Ct values of 22.9–35.4 (n = 12), only a few reads could be mapped to the PRRSV genome, and no full-length genomic sequences were obtained from these samples.
Discussion
There are several advantages of first attempting NGS on virus isolates: 1) PRRSV isolates grown in cell cultures are relatively “pure” with fewer host gene sequences; 2) abundant PRRSV genetic material is present in the cell culture–derived isolates; 3) starting with PRRSV isolates can help to optimize procedures for sequencing and analysis; and 4) inclusion of some PRRSV reference isolates with known sequences can confirm the integrity and quality of the sequence data obtained by NGS technology. In the current study, 10 well-characterized PRRSV isolates were hence first used to optimize the NGS procedures. Whole genome sequences of all 10 PRRSV reference isolates were successfully obtained using NGS. For these PRRSV isolates, the nucleotide identities between the NGS sequences and the previously reported sequences were 99.3–99.9%. It is not surprising that the nucleotide identities were not 100% because the virus isolates used for sequencing in our study may not have the same passage history as the isolates whose sequences have been deposited in GenBank.
A key question was whether NGS technology can detect and distinguish a mixed infection with 2 PRRSV strains. We found that, by using routine sequence data analysis procedures (mapping sequence reads to reference PRRSV genome library followed by de novo assembly), NGS was able to detect and distinguish a mixture containing a PRRSV-1 Lelystad strain and a PRRSV-2 VR-2385 strain (these 2 strains have 57.8% nucleotide identity at the whole genome level) with their respective full genomic sequences obtained. However, for a mixture containing a PRRSV-2 vaccine strain (the MLV vaccine a ) and a PRRSV-2 VR-2385 strain (these 2 strains have 92.4% nucleotide identity at the whole genome level), routine sequence analysis procedures including de novo assembly could obtain the consensus of 2 genome sequences but could not obtain separately assembled genome sequences for 2 viruses. Use of another bioinformatics program (i.e., IGV software) showed nucleotide variations at each single position of sequence reads and enabled us to demonstrate the presence of viral quasi-species or more than 1 PRRSV strain. Nevertheless, the IGV software could not automatically distinguish the MLV vaccine from the VR-2385 sequence reads to assemble into their respective full genomic sequences. If at least 1 PRRSV isolate in the mixture has known genome sequences, it may be possible to manually determine whether a nucleotide at one position belongs to this PRRSV isolate or not. Generally speaking, intratypic pairwise nucleotide sequence variations are up to 30% among PRRSV-1 viruses and more than 21% in PRRSV-2 viruses, respectively, based on ORF5 sequences. 47 The MLV vaccine and VR-2385 viruses evaluated in our study have ~7% nucleotide differences. Based on the current study data, we cannot definitively conclude that NGS cannot distinguish and obtain genome sequences of two PRRSV-1 viruses or two PRRSV-2 viruses in a mixture; the outcomes may depend on how different the 2 respective viruses are in a mixture. This area remains to be further investigated.
From the laboratory and swine industry points of view, it is important to establish an efficient procedure to determine whole genome sequences directly from clinical samples. Serum, oral fluid, and lung samples are currently the 3 most common specimen types submitted by swine veterinarians for PRRSV PCR testing. Therefore, serum, lung, and oral fluid were chosen as clinical specimen types in our study to evaluate the performance of NGS technology in determining whole genome sequences of PRRSV. Full-length genomic sequences were successfully obtained from serum samples with Ct values 15.1–23.6, lung samples with Ct values 16.4–21, and oral fluid samples with Ct values 18.7–20.6. When Ct values were ≥24.3 for serum samples, ≥21.3 for lung samples, and ≥22.9 for oral fluids, full-length sequences could not be obtained, with assembly of no or only partial contigs possible. This implies that different specimen types evaluated in this study did not markedly affect the NGS efficiency; instead the Ct value of PRRSV in a sample is inversely related to the NGS outcomes. In addition to Ct values of PRRSV, the sample quality and PRRSV RNA integrity could also be potential factors affecting NGS outcomes on various samples. The RNA integrity number (RIN) determined by a bioanalyzer has generally been used to estimate the integrity of eukaryote and bacterial total RNA. However, it has been a challenge to accurately and specifically assess viral RNA integrity because viruses generally lack rRNA.
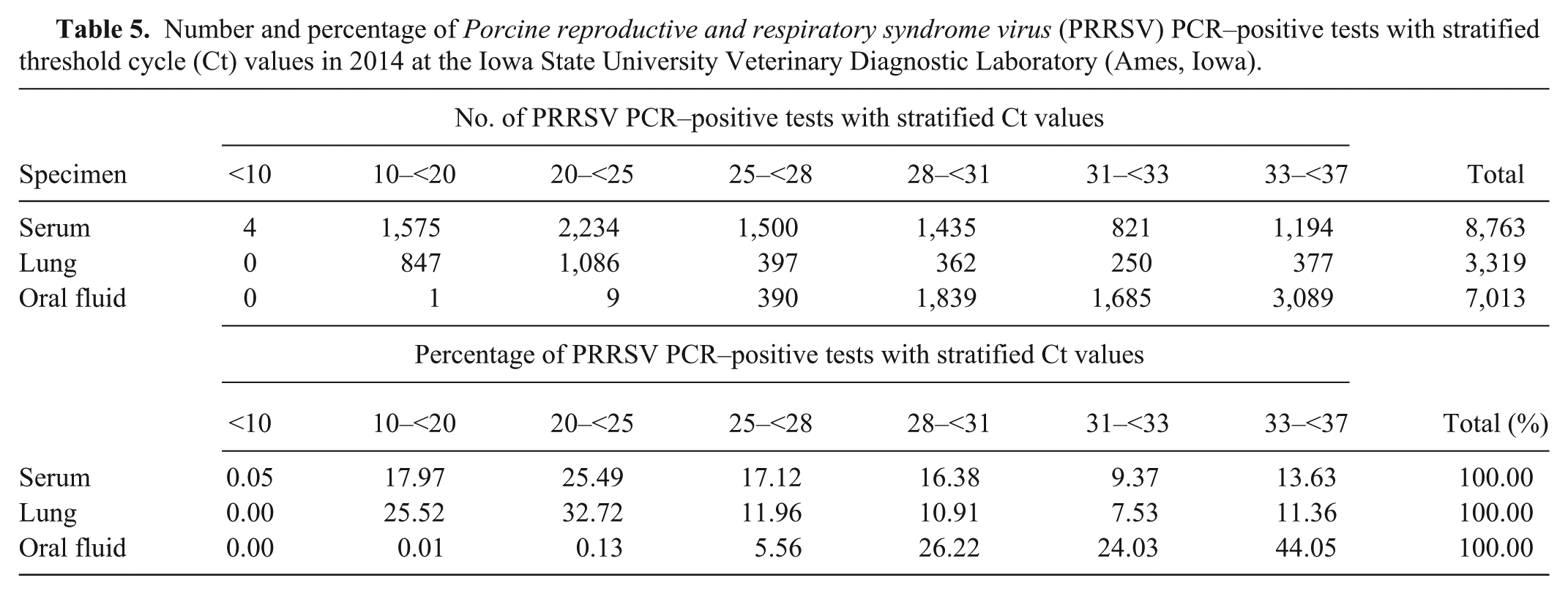
The NGS data obtained in our study together with PRRSV PCR–positive tests (Table 5) suggest that ~43% of PRRSV PCR–positive serum samples submitted to ISU VDL in 2014 had Ct <25 and would likely undergo successful whole genome sequencing by NGS; ~40% of PRRSV PCR–positive lung samples had Ct <22 and would likely undergo successful full-length genome sequencing by NGS; ~0.14% of PRRSV PCR–positive oral fluids had Ct <25 and would likely undergo successful whole genome sequencing by NGS. We do not recommend performing NGS on clinical serum, lung, and oral fluid samples with Ct >25 in the expectation of obtaining full-length PRRSV genomic sequences from these samples.
Number and percentage of Porcine reproductive and respiratory syndrome virus (PRRSV) PCR–positive tests with stratified threshold cycle (Ct) values in 2014 at the Iowa State University Veterinary Diagnostic Laboratory (Ames, Iowa).
Using the traditional Sanger method, all serum, lung, and oral fluid samples with Ct <31 had >95% success rates in PRRSV ORF5 sequencing; for serum, lung, and oral fluid samples with Ct 31–<33, the ORF5 sequencing success rates were 92.1%, 76.1%, and 86.7%, respectively; for serum, lung, and oral fluid samples with Ct 33–<37, the ORF5 sequencing success rates were 78.8%, 29.8%, and 57.2%, respectively (Table 6). Thus, for clinical serum, lung, and oral fluid samples with Ct 25–37, NGS may not be a good choice for whole genome sequencing but ORF5 sequencing using the Sanger method would have fairly good success rates on these samples and could be an option for swine practitioners, diagnosticians, and researchers.
Correlation of Porcine reproductive and respiratory syndrome virus (PRRSV) real-time reverse transcription PCR threshold cycle (Ct) values to ORF5 Sanger sequencing (Seq) outcome based on specimen types.*
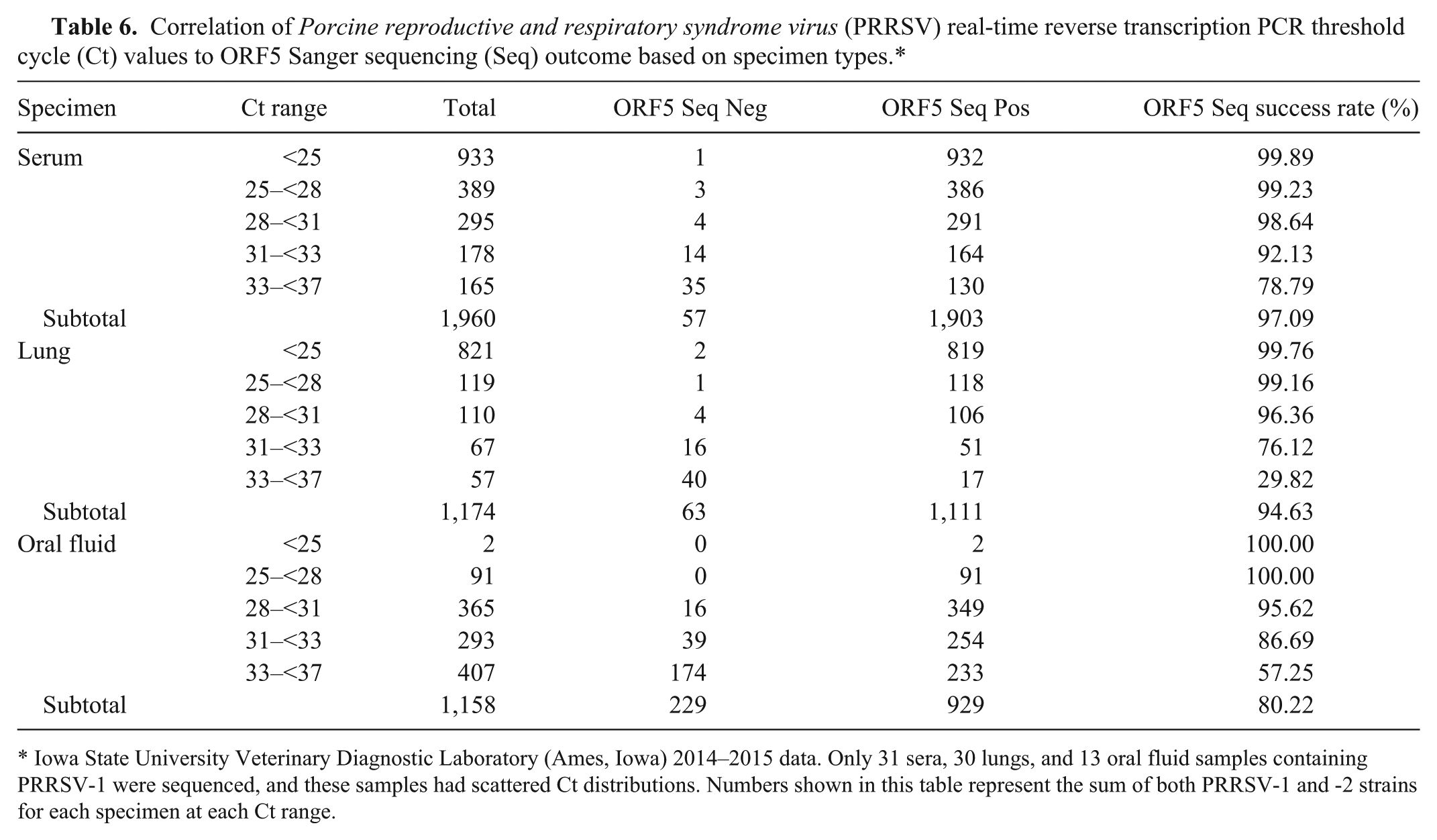
Iowa State University Veterinary Diagnostic Laboratory (Ames, Iowa) 2014–2015 data. Only 31 sera, 30 lungs, and 13 oral fluid samples containing PRRSV-1 were sequenced, and these samples had scattered Ct distributions. Numbers shown in this table represent the sum of both PRRSV-1 and -2 strains for each specimen at each Ct range.
It must be noted that, in our study, 24 samples were multiplexed in a single NGS sequencing reaction. If fewer samples are multiplexed in a single NGS sequencing reaction, the success rate of obtaining the PRRSV whole genome sequences from clinical specimens using NGS MiSeq system could be higher, but the cost per sample would increase accordingly. Another limitation of the current study is that only 51 serum, lung, and oral fluid samples with different ranges of PRRSV PCR Ct values were tested by NGS; testing more clinical samples in each Ct range would increase statistical power and draw a stronger conclusion on the relationship between Ct values and NGS success rate. This warrants further investigation in future studies.
We developed an efficient and streamlined NGS approach to quickly determine the complete genomic sequences of PRRSV in cell culture–derived isolates and clinical specimens. Our method does not require sequence-specific primers for PCR amplification or bait-based enrichment as described previously, 17 thus limiting the possibility of primer-based bias in the processing of samples and providing the opportunity of detecting any PRRSV variants at the same time. Our strategy is also different from that described in some previous studies,22,23 which relies on bioinformatic filtering of host reads. In our NGS approach, total RNA is directly extracted from virus isolates and/or clinical specimens, and the cDNA libraries of different samples can be indexed and simultaneously sequenced, making it an efficient approach for use in diagnostic investigations and research. Regarding data analysis, previous methods of reference mapping usually used 1 reference genome, 2 which is restricted to the samples with known virus strains. However, considering the high genetic diversity among PRRSV strains, a single reference genome might not be similar enough for mapping reads, and consensus sequences may not be constructed successfully using SAMtools mpileup command. 21 Therefore, we built a reference genome library containing all of the PRRSV genome sequences publicly available to extend the range of mapping objects. Usually, more reads can be mapped onto the reference genome library as compared to mapping against a single genome, and thereby more reads of viruses of interest can be collected for de novo assembly. The approach described in the current study has also been used to successfully obtain the whole genome sequences of other RNA viruses, such as Porcine epidemic diarrhea virus, porcine deltacoronavirus, Equine arteritis virus, and Senecavirus A, in forms of cell culture–derived isolates or clinical specimens with high concentrations of viruses.7,8,19,29,46 However, the success rate of determining the whole genome sequences of PRRSV and other viruses from clinical samples with high Ct values was not satisfactory using our current NGS approach. Additional work remains to improve success rates of whole genome sequencing using NGS technology on clinical samples with low viral concentrations.
Footnotes
Acknowledgements
We thank Drs. Chandrasen Soans, Michael Bishop, and others from Illumina for technical training on next-generation sequencing.
Authors’ note
Jianqiang Zhang and Ying Zheng contributed equally to this work.
Authors’ contributions
J Zhang contributed to conception and design of the study; contributed to analysis and interpretation of data; drafted the manuscript; critically revised the manuscript; and gave final approval. Y Zheng contributed to acquisition and analysis of data, and drafted the manuscript. XQ Xia contributed to analysis of data. Q Chen and SA Bade contributed to acquisition of data and critically revised the manuscript. KJ Yoon contributed to conception of the study; critically revised the manuscript; and gave final approval. KM Harmon, PC Gauger, and RG Main critically revised the manuscript and gave final approval. G Li contributed to conception and design of the study; contributed to acquisition, analysis, and interpretation of data; drafted the manuscript; critically revised the manuscript; and gave final approval. All authors agreed to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
MLV vaccine: Ingelvac PRRS MLV, Boehringer Ingelheim Vetmedica, St. Joseph, MO.
b.
ATP vaccine: Ingelvac PRRS ATP, Boehringer Ingelheim Vetmedica, St. Joseph, MO.
c.
F vaccine: Fostera PRRS, Zoetis, Florham Park, NJ.
d.
VetMax NA and EU PRRSV reagents, Thermo Fisher Scientific, Waltham, MA.
e.
Earle’s balanced salt solution, Sigma-Aldrich, St. Louis, MO.
f.
Phosphate buffered saline (PBS, 1× pH 7.4), Thermo Fisher Scientific, Waltham, MA.
g.
Kingfisher-96 magnetic particle processor, Thermo Fisher Scientific, Waltham, MA.
h.
MagMAX-96 viral RNA isolation kit, Thermo Fisher Scientific, Waltham, MA.
i.
Total RNA purification kit, Norgen Biotek, Thorold, Ontario, Canada.
j.
Qubit 2.0 spectrophotometer, Thermo Fisher Scientific, Waltham, MA.
k.
Qubit RNA BR (Broad-Range) assay kit, Thermo Fisher Scientific, Waltham, MA.
l.
TruSeq stranded total RNA sample preparation kit, Illumina, San Diego, CA.
m.
Agencourt RNAClean XP, Beckman Coulter, Brea, CA.
n.
Agilent 2100 Bioanalyzer, Agilent Technologies, Santa Clara, CA.
o.
Agencourt AMPure XP, Beckman Coulter, Brea, CA.
p.
KAPA library quantification kit, KAPA Biosystems, Wilmington, MA.
q.
Illumina MiSeq sequencer, Illumina, San Diego, CA.
u.
DNASTAR Lasergene 11 Core Suite, DNASTAR, Madison, WI.
v.
MagMAX pathogen RNA/DNA kit, Thermo Fisher Scientific, Waltham, MA.
w.
ABI 7500 Fast instrument, Thermo Fisher Scientific, Waltham, MA.
x.
qScript custom one-step RT-PCR kit, Quanta Biosciences, Gaithersburg, MD.
y.
QIAxcel advanced system, Qiagen, Valencia, CA.
z.
ExoSAP-IT kit, Affymetrix, Santa Clara, CA.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by funds from Zoetis and the Iowa State University Veterinary Diagnostic Laboratory. Illumina provided in-kind support of some reagents.