
Abstract
In an effort to improve a competitive blocking enzyme-linked immunosorbent assay (cELISA) for antibody detection to Equine arteritis virus (EAV), antigen purified by anion-exchange membrane chromatography capsule (AEC) was evaluated. Virus purification by the AEC method was rapid and easily scalable. A comparison was made between virus purified by the AEC method with that obtained by differential centrifugation based on the following: 1) the relative purity and quality of EAV glycoprotein 5 (GP5) containing the epitope defined by monoclonal antibody 17B7, and 2) the relative sensitivity of a commercial antibody cELISA with the only change being the 2 purified antigens. On evaluation by Western blot using GP5-specific monoclonal antibody 17B7, the AEC-purified EAV contained 86% GP5 monomer whereas the differentially centrifuged EAV contained <29% of the monomer. Improvement of analytical sensitivity without sacrifice of analytical specificity was clearly evident when cELISAs prepared with EAV antigen by each purification method were evaluated using 7 sensitivity and specificity check sets. Furthermore, the AEC-purified EAV–based cELISA had 30–40% higher agreement with the virus neutralization (VN) test than the cELISA prepared with differentially centrifuged EAV based on testing 40 borderline EAV-seropositive samples as defined by the VN test. In addition, the AEC-purified cELISA had highly significant (P = 0.001) robustness indicated by intra-laboratory repeatability and interlaboratory reproducibility when evaluated with the sensitivity check sets. Thus, use of AEC-purified EAV in the cELISA should lead to closer harmonization of the cELISA with the World Organization for Animal Health–prescribed VN test.
Keywords
Introduction
A number of ion-exchange membrane chromatographic methods have been successfully used for the purification of several viruses, including adenoviruses, herpesviruses, influenza viruses, and densonucleosis virus.15,17,18,22 Chromatography based on membrane materials and chemistries has several advantages in bioseparation and downstream processing of large complex biomolecules, including viruses, over chromatographic methodologies based on other materials, such as resins.3,14,22 Simple and rapid membrane chromatography–based purification of killed or live vaccines improve their quality by reducing unwanted or unnecessary components in vaccine formulation.17,22 Moreover, ion-exchange membrane chromatography techniques also may be useful for purification of viruses and viral antigens used in diagnostic tests. In our report, ion-exchange membrane chromatography methods were evaluated for purification of Equine arteritis virus (EAV), and an anion-exchange membrane chromatography capsule (AEC) method was selected for further investigation. The AEC-purified EAV was compared with EAV purified by differential centrifugation using a commercial competitive blocking enzyme-linked immunosorbent assay (cELISA) and other analyses.
Equine arteritis virus (order Nidovirales, family Arteriviridae, genus Arterivirus) is the causative agent of equine viral arteritis (EVA). Exposure to EAV may result in clinical or inapparent infection. Clinical signs of EVA can include fever, anorexia, conjunctivitis, nasal discharge, dependent edema, abortion, and infrequently, death in young foals.2,25 Transmission by acutely infected horses is usually through direct contact with respiratory secretions containing EAV. An economically challenging consequence of infection is that 30–70% of naive stallions can become clinically inapparent carriers following recovery from the acute phase of the infection. 26 Therefore, careful screening and early identification of carrier stallions using sensitive and specific diagnostic tests are critical to the effective control of EAV on breeding farms. Various ELISAs have been developed to detect EAV antibody,4,6,9,12 but none have been widely validated for field use. 27 The virus neutralization (VN) test principally detects antibodies to the immunodominant glycoprotein 5 (GP5), 5 and is considered the most sensitive assay to detect EAV-specific antibodies in horse serum (Senne et al., Equine viral arteritis: a standard procedure for the virus neutralization test and comparison of results of a proficiency test performed at 5 laboratories, Proceedings of the 89th US Animal Health Association meeting, October 1985, Milwaukee, Wisconsin). The VN test, which involves a test time of 72 hr, is currently the prescribed test for international trade by the World Organization for Animal Health (OIE), and a VN antibody titer of ≥1:4 is considered positive evidence of infection with EAV. 27 However, variation exists between some laboratories in performance of the VN test. 27 This makes global harmonization of EAV testing challenging in spite of continued efforts by OIE and its Reference Laboratories to persuade laboratories to adhere to the test protocol detailed in the relevant OIE Manual 27 and to use a common standard. This emphasizes the importance of developing a more robust, faster, and user-friendly assay that is closely harmonized with the VN test. It is critical that there is a high level of concurrence between both assays in correctly identifying sera at the cutoff dilution of 1:4. The availability of such an assay would be a significant contribution to helping countries prevent and control outbreaks of EVA.
An improved cELISA based on an EAV GP5 nonneutralizing epitope was previously reported with a diagnostic specificity of 99.8% and sensitivity of 95.5% when compared with VN test results, respectively. 7 In a field trial conducted with a number of American Association of Veterinary Laboratory Diagnosticians–accredited state laboratories and an OIE Reference Laboratory for EVA, the diagnostic specificity of the same cELISA was 99.5% and the diagnostic sensitivity was 98.2%. 8 The cELISA was not adversely affected by previous exposure of horses to non–EAV-containing biologicals,6–8 which caused problems in an indirect ELISA and the VN test due to the antibodies generated in horses against antigens in the RK-13 (rabbit kidney) cell line used in vaccine production.6,9 Since introduction of the commercial cELISA for EAV antibody determination, some sera with low VN-positive titers were found to be negative by cELISA (Pfahl et al., Evaluation of a commercially available ELISA (cELISA) for the detection of antibodies to Equine arteritis virus (EAV), Proceedings of the 47th AAVLD Meeting, October 2014, Kansas City, Missouri). Our study describes efforts to address this problem through further enhancement in the sensitivity of the improved cELISA using EAV antigen purified by AEC with the goal of providing a simpler, more robust, and rapid serological assay for EAV.
Materials and methods
Propagation of EAV
The Bucyrus strain of EAV (Edwards et al., International harmonisation of laboratory diagnostic tests for equine viral arteritis, Proceedings of the 8th International Conference on Equine Infectious Diseases, March 1998, Dubai, UAE) was propagated in adventitious virus–free primary equine spleen (ESP) cells. Confluent cell monolayers in 850-cm2 roller bottle flasks were inoculated with a multiplicity of infection of ~0.0005. After incubation for 2–3 days at 37°C in serum-free Eagle minimal essential medium (EMEM) a or EMEM containing 5% fetal bovine serum (FBS), b and when virus-induced cytopathic effect had reached a maximum, cells and culture supernatant were combined and centrifuged at 4,000 × g for 10 min at 4°C to eliminate cell debris.
Titration of EAV
Equine arteritis virus–containing cell culture supernatant and fractions from the AEC purification of EAV were titrated, and the 50% tissue culture infective dose (TCID50) was calculated according to the method of Reed and Muench. 23 Briefly, cultures of ESP cells in 96-well plates were inoculated with serial 10-fold dilutions of EAV suspension using EMEM containing 5% FBS. The development of EAV cytopathic effect in the ESP cell wells was recorded daily for 7 days and the results used to calculate the TCID50/mL.
Purification of EAV using differential centrifugation method
Purification of EAV using differential centrifugation was performed according to a previously described method with some modifications. 6 Briefly, the virus-containing supernatant was centrifuged at 21,000 × g for 18 hr at 4°C. The supernatant was discarded, and the virus-containing pellet was resuspended to 1/50 of its original volume in phosphate buffered saline (PBS; pH 7.2) and centrifuged at 4,000 × g for 10 min at 4°C. The resulting supernatant (S1) was saved. The pellet was again resuspended in PBS to 1/50 of its original volume and centrifuged at 4,000 × g for 10 min at 4°C. The resulting supernatant (S2) and S1 were combined and subjected to ultracentrifugation at 45,000 × g for 4 hr at 4°C. After centrifugation, the supernatant was discarded, and the virus-containing pellet was resuspended in 0.85% saline (pH 7.2) to 1/1,000 of its original volume. Triton X-100 c at a final concentration of 0.2% was added, and the mixture was stirred overnight at 4°C followed by 1 hr at 37°C. The solubilized preparation was centrifuged at 15,000 × g for 10 min at 4°C, and the resultant supernatant was used as coating antigen in the cELISA.
Purification of EAV using ion-exchange membrane capsules
Equine arteritis virus propagated in ESP cells cultured in serum-free EMEM or EMEM containing 5% FBS was used in the purification of EAV using 2 types of ion-exchange membrane capsules, namely, AEC and cationic exchange capsule. d Briefly, the membrane capsules were primed with 10 mL of 1 M NaCl, and then 10 mL of equilibrium buffer (EMEM diluted 2-fold in sterile distilled water) was pumped through the capsule using a peristaltic pump with a flow rate of 50–200 mL/min. Two hundred milliliters of EAV culture supernatant diluted 2-fold in sterile distilled water was passed through the capsule with a flow rate of 50–200 mL/min. Then, the capsule was rinsed with 10 mL of equilibrium buffer. The EAV bound in the membrane capsule was eluted with 10 mL of NaCl solutions (0.3, 0.6, and 1.1 M). The eluted fractions were collected and evaluated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and silver staining as well as for EAV antigen by Western blot analysis using EAV GP5-specific monoclonal antibody (mAb) 17B7. In addition, the fractions resulting from the procedure were monitored for infective EAV by determining their TCID50/mL.
Test equine sera
Four sensitivity check sets were prepared by diluting EAV-positive sera in negative equine serum: 1:16–1:128 dilutions of serum SR10387, 1:256–1:16,384 dilutions of serum H537, 1:8–1:128 dilutions of serum H631, and 1:4–1:128 dilutions of serum H632. Another check set was prepared with sequentially collected sera (collection every 12 hr for 5–9 days post–modified live vaccine [MLV] vaccination) from horse H681. Horse SR10387 was an EAV carrier, and the remaining 4 horses (H537, H631, H632, and H681) were vaccinated with the MLV against EVA. e The EAV VN antibody status of each sample in these 5 sensitivity check sets was determined. These samples were used to compare the analytical sensitivity and specificity, intra-laboratory repeatability, and inter-laboratory reproducibility of the cELISAs. A sixth sensitivity check set was also included: 40 EAV VN antibody–positive sera, most of which gave borderline VN test results that were close to the ≥1:4 cutoff value. Sera giving borderline VN test–positive results were collected immediately after seroconversion and were used to compare the cELISAs developed with EAV purified by the 2 purification methods. A seventh serum panel included 38 sera with high negative cELISA readings (>10% inhibition, but <35% inhibition) and 45 sera with negative results in VN test (<1:4); the cELISA results were determined with a kit developed with EAV purified by differential centrifugation. These 38 sera negative in both VN test and commercial cELISA were included as a true-negative check set for initially evaluating the diagnostic specificity of the cELISA based on AEC-purified EAV.
Competitive blocking ELISA
The cELISA used in our study was described in detail previously.6–8 A dilution of EAV antigen that gave an optical density (OD) reading of ~0.800 at 450 nm (A450) when tested against the negative reference serum was made in 0.05 M carbonate buffer (pH 9.6). This was used to coat high-binding 96-well plates, f adding 50 µL/well. The plates were sealed and incubated for 2 hr at 37°C, before adding 200 µL/well of blocking buffer. g After blocking, the plates were dried overnight at 25°C, and stored individually in bags h at 4°C.
Stored plates and all other reagents used in the cELISA procedure were warmed to room temperature (23 ± 2°C) before 50 µL/well of test sera were added to the antigen-coated plates. The plates were incubated for 2 hr and then washed; an optimized dilution of mAb 17B7 in antibody dilution buffer i was added to each well, and the plates were incubated for 30 min before washing. An optimized dilution of goat anti-mouse immunoglobulin G conjugated to horseradish peroxidase j in antibody dilution buffer was added to each well, and the plates were re-incubated for 30 min. After incubation and washing, tetramethylbenzidine substrate solution k was added to each well. After incubation for 15 min, the reaction was stopped by the addition of stop solution, l and the OD at A450 was determined. m The percent inhibition (%I) of mAb 17B7 binding by the test serum was calculated using the following formula: %I = 100 – [(test serum OD × 100) ÷ (mean negative control OD)]. The test serum was considered to be antibody-positive if the %I was ≥35%.
Virus neutralization test
The OIE-prescribed VN test, which is a complement-enhanced microtiter assay for measuring EAV-neutralizing antibodies in equine serum, was performed as previously described (Senne et al., Equine viral arteritis).
Western blot analysis of EAV using anti-GP5 monoclonal antibody
To evaluate the quality of EAV GP5 prepared by the 2 different purification methods, Western blot analysis was performed with GP5-specific mAb 17B7 n according to the previously described method. 11 Briefly, purified EAVs were adjusted to contain the same protein concentration, and boiled for 3 min in sample buffer o containing fresh 2-mercaptoethanol followed by SDS-PAGE. p Then, the SDS-PAGE gels with AEC-purified and centrifugation-purified EAVs were stained and compared for a broad range of proteins using a silver staining kit. q For Western blot analysis, EAVs in SDS-PAGE gels were transferred to nitrocellulose membranes and the membranes blocked with Tris–Tween-20 buffer containing 5% skim milk. The GP5-containing bands were detected with EAV GP5-specific mAb 17B7 followed by treatment with peroxidase-conjugated goat anti-mouse immunoglobulin G. r The relative quantity of Western blot–detected bands in EAV purified by AEC or differential centrifugation was analyzed with ImageJ software. 24 Based on the net density analyzed on ImageJ, a comparison of the relative quantity of GP5 monomer, GP5-M heterodimer, and other macromolecules associating with GP5 in the EAV fractions after SDS-PAGE under reducing conditions was carried out.
Statistical analysis
Both parametric t-tests and nonparametric Wilcoxon tests were used to determine if there were significant (P < 0.05) differences between cELISAs using AEC- and differential centrifugation–purified EAV. 1 In addition, the inter-run and inter-laboratory variability in testing with the cELISA using AEC-purified EAV were also analyzed using the same 2 methods. All statistical analyses were performed using R software (http://www.r-project.org/).
Results
Optimization of AEC purification of EAV
Two different types of membrane chromatography including strong cationic and strong anionic exchange capsules were compared for EAV capture capacity using the same volume (250 mL) and titer (106 TCID50/mL) of EAV-containing culture supernatant from ESP cells grown in EMEM supplemented with FBS. Based on these initial experiments, the procedure using a strong AEC with pH 7.2 buffer had better capture and elution of EAV than a strong cation-exchange membrane capsule. This was based on the AEC method removing more EAV infectivity from the culture supernatant determined by virus titration and by the amount of EAV GP5 detected by Western blot analysis of eluted EAV fractions using GP5-specific mAb 17B7 (Fig. 1). For further optimization of EAV capture capacity by AEC, EAV propagation was carried out in serum-free medium to reduce total protein in the supernatant, and 1.75 L of this low-protein EAV culture supernatant was used for AEC purification. After passage through an AEC capsule, no infectious EAV was detected in the fall-through and the washing buffer (the detection limit of the EAV infectivity assay was101.75 TCID50/mL). This indicated that the AEC capsule captured >99.9% (>109 infectious virus particles) of the EAV in the suspension added to the capsule. The eluted fractions were not evaluated for infectious EAV because of likely inactivation by the NaCl solutions used for elution.

Western blot of Equine arteritis virus (EAV) antigen eluted from 2 types of ion-exchange membrane capsules. Lanes A–C contain cation exchange c elutions 1–3, respectively, whereas lanes D–F contain anion-exchange c elutions 1–3, respectively. The same volume of elution buffer was used to elute EAV for both purifications, and the same volume of sample was loaded onto sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Difference in diagnostic performance between cELISAs developed with EAVs purified by 2 different methods
To determine if AEC-purified EAV improved the analytical sensitivity of the cELISA, separate kits were produced using AEC-purified and differentially centrifuged EAVs, respectively. In the comparison testing using 3 sensitivity check sets prepared by serially diluting sera from MLV-vaccinated horses (H537, H631, H632) in an EAV-negative serum, the cELISA kit prepared with AEC-purified EAV had a 2- to 4-fold higher analytical sensitivity than the corresponding cELISA prepared with EAV purified by differential centrifugation (Table 1). On analysis of the results following testing of the check set made with sequentially collected sera from a MLV-vaccinated horse (H681), the cELISA based on AEC-purified EAV detected EAV antibody 24 hr earlier and had significantly (P = 0.001) higher %I at all tested time points compared to the cELISA based on EAV purified by differential centrifugation (Fig. 2). The results were repeatable in 6 runs within the 1 laboratory without significant variability (P = 0.2, average standard deviation 2.7; Fig. 3). The results were also reproducible in runs between 2 independent laboratories without significant variability (P = 0.2, average standard deviation 3.2; Table 1). The difference in analytical sensitivity was 1 two-fold dilution, and the average %I difference between the 2 cELISAs were 16.7, 16.3, 12.2, and 12.2 %I when tested using 1:16–1:128 two-fold serial dilutions of SR10387 serum from an infected carrier horse, respectively (Fig. 3).
Comparison of the analytical sensitivity of competitive blocking enzyme-linked immunosorbent assays (cELISA) made with Equine arteritis virus (EAV) antigen purified by 2 different methods with the virus neutralization (VN) test.*
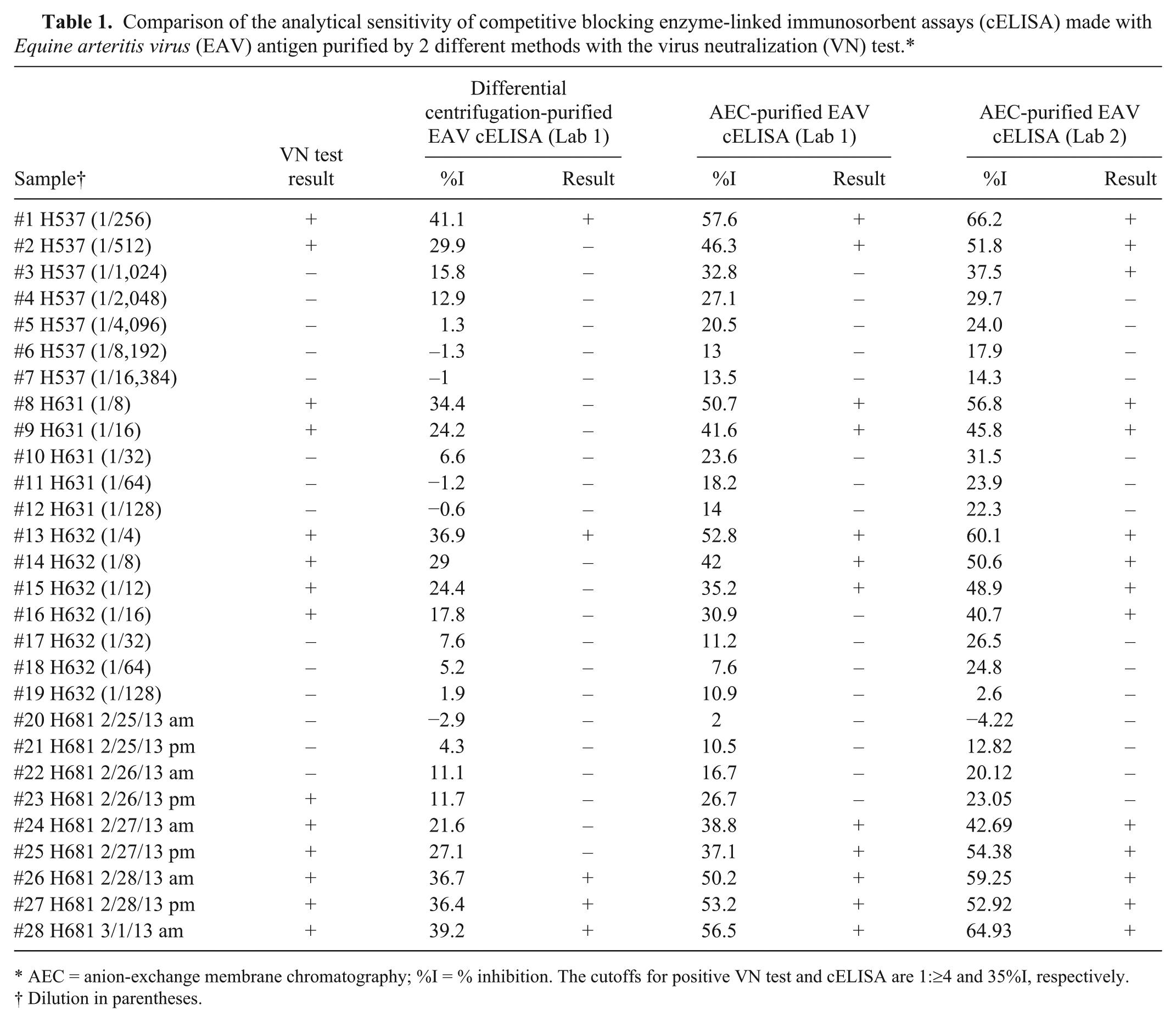
AEC = anion-exchange membrane chromatography; %I = % inhibition. The cutoffs for positive VN test and cELISA are 1:≥4 and 35%I, respectively.
Dilution in parentheses.
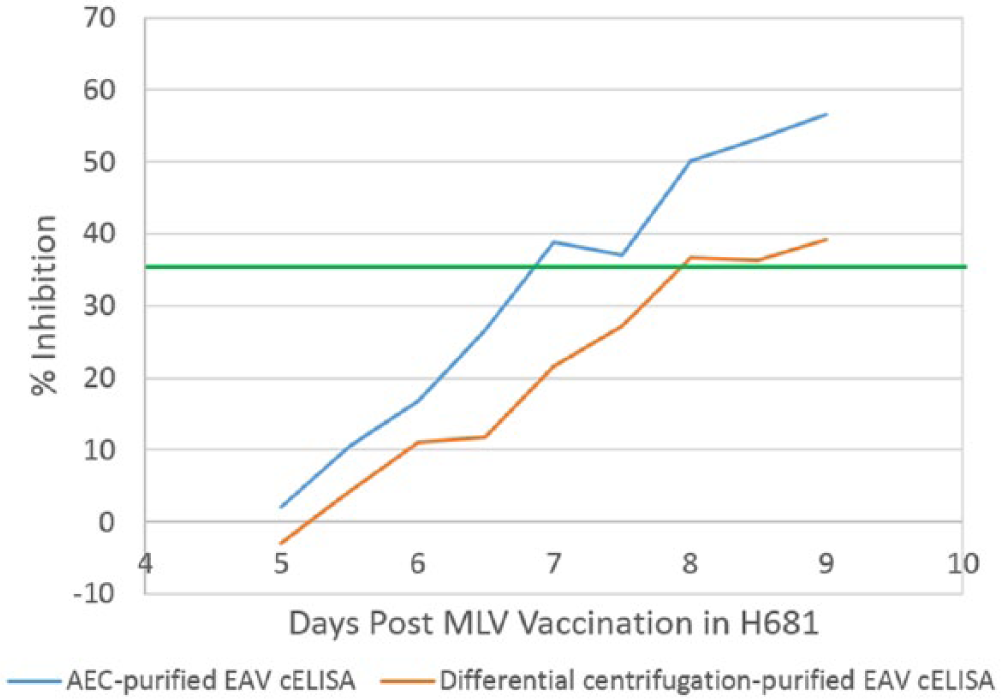
Comparison between the competitive blocking enzyme-linked immunosorbent assays (cELISA) made with anion-exchange membrane chromatography (AEC)-purified and differential centrifugation–purified Equine arteritis virus (EAV) using sera collected after vaccination with an EAV modified live vaccine (MLV): green line represents the cutoff (35% inhibition) for positive and negative results.
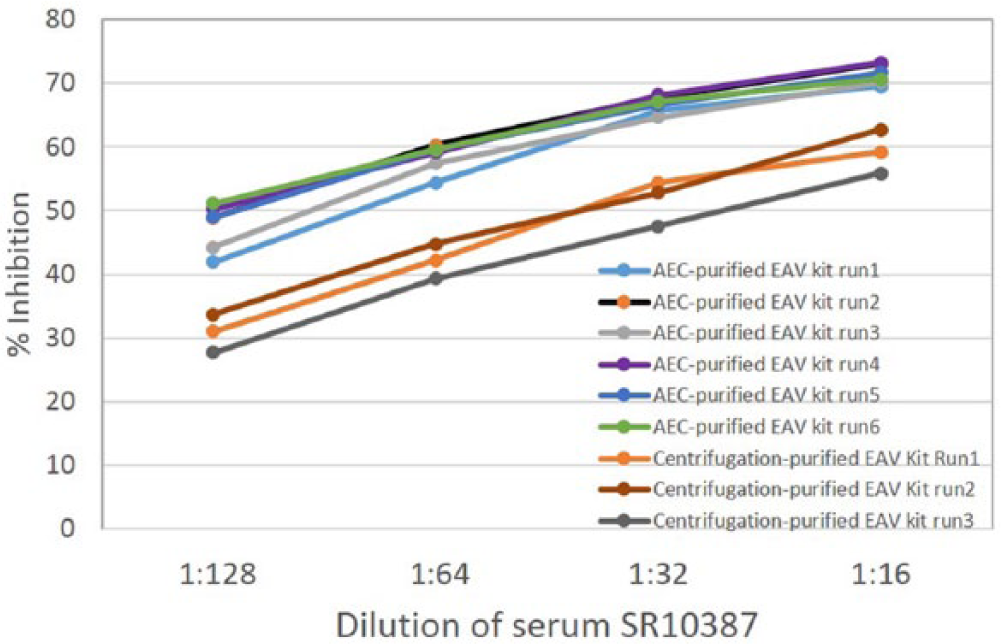
Repeatability of analytical sensitivity difference between the competitive blocking enzyme-linked immunosorbent assays made with anion-exchange membrane chromatography (AEC)-purified and differential centrifugation–purified Equine arteritis virus (EAV).
Forty horse sera with VN titers ranging between 1:4 and 1:64, in terms of the median titer of the results from up to 4 repeat VN tests, were defined as a borderline diagnostic sensitivity check set. The variability in VN titers of the 40 sera when tested by an experienced operator in 2–4 separate runs was up to 4-fold as presented in high standard deviation of VN test titers determined from up to 4 separate runs (Fig. 4A). Six of the 40 sera had 1 negative VN test result (<1:4 titer), but the other 3 replicates were positive for all 6 sera with titers ranging from 1:4 to 1:8 (Fig. 4A). These 40 borderline positive sera were analyzed with the 2 different cELISAs prepared with EAV purified using AEC or differential centrifugation. Using the cELISA developed with AEC-purified EAV, the results were 82.5% in agreement with the VN test results (Table 2; Fig. 4B). However, the cELISA kit lots made with EAV purified by differential centrifugation were only 32.5% in agreement with the VN test results (Table 2; Fig. 4C).
Diagnostic performance of competitive blocking enzyme-linked immunosorbent assays (cELISA) made with Equine arteritis virus (EAV) purified by 2 different methods with borderline virus neutralization (VN) test–positive sera. For each assay (VN test; cELISA with anion-exchange membrane chromatography (AEC)-purified EAV antigen; and cELISA with differential centrifugation–purified EAV antigen), the mean VN titer or the % inhibition based on 3 repeats was used to calculate the standard deviation (SD). The cELISA repeats were conducted using different kit lots. The mean % inhibition or the VN titer and the standard deviation of each tested sample are presented in graphs with the error bar.
Diagnostic agreement of competitive blocking enzyme-linked immunosorbent assays (cELISA) made with Equine arteritis virus (EAV) purified by 2 different methods with borderline virus neutralization (VN) test–positive sera.*

The borderline positive sera were selected based on the results in 2–4 separate VN tests by an experienced operator. AEC = anion-exchange membrane chromatography. 1 Number of samples with agreement between each cELISA and VN test results. Numbers in parentheses are % correlation.
The mean % inhibition of each sample determined based on 3 runs using different kit lots was used to calculate the number of positive samples and % correlation against VN test results.
The mean VN titer of each sample was used to calculate the number of positive samples: 2 samples had a mean VN titer <1 (Log 2) when determined with 4 separate VN test runs.
In spite of significantly improved sensitivity of the AEC-purified EAV cELISA, there was no loss in specificity following testing of 38 sera that were negative by VN test and a previous cELISA prepared with differentially centrifuged EAV (Table 3). The test agreement between the AEC-purified EAV cELISA and the differential centrifugation–based EAV cELISA was 100% when tested with 1 lot of AEC-purified EAV cELISA and 2 lots of the differential centrifugation–purified EAV cELISA (Table 3). Further testing of 15 sera available with enough sample volumes gave 100% in specificity when tested in 2 additional lots of AEC-purified EAV cELISA (see Supplemental Table 1 available at http://vdi.sagepub.com/content/by/supplemental-data). In an additional evaluation of the AEC-purified EAV cELISA using 45 sera with negative VN test results, all 3 kit lots (lot 423, 423a, and P1304) had 100% performance (all negative results, ≤30% inhibition) with a resolution comparable to the differential centrifugation–based EAV cELISA: −13 and −27.6 to 0.5, −5.5 and −17.6 to 8.1, 0.39 and −11.9 to 10.2 in mean %I and %I range of 3 lots (lot 423, 423a, and P2014), respectively (see Supplemental Table 2 available at http://vdi.sagepub.com/content/by/supplemental-data).
Comparison in diagnostic specificity between competitive blocking enzyme-linked immunosorbent assays (cELISA) made with Equine arteritis virus (EAV) purified by 2 different methods with virus neutralization (VN) test–negative sera.*
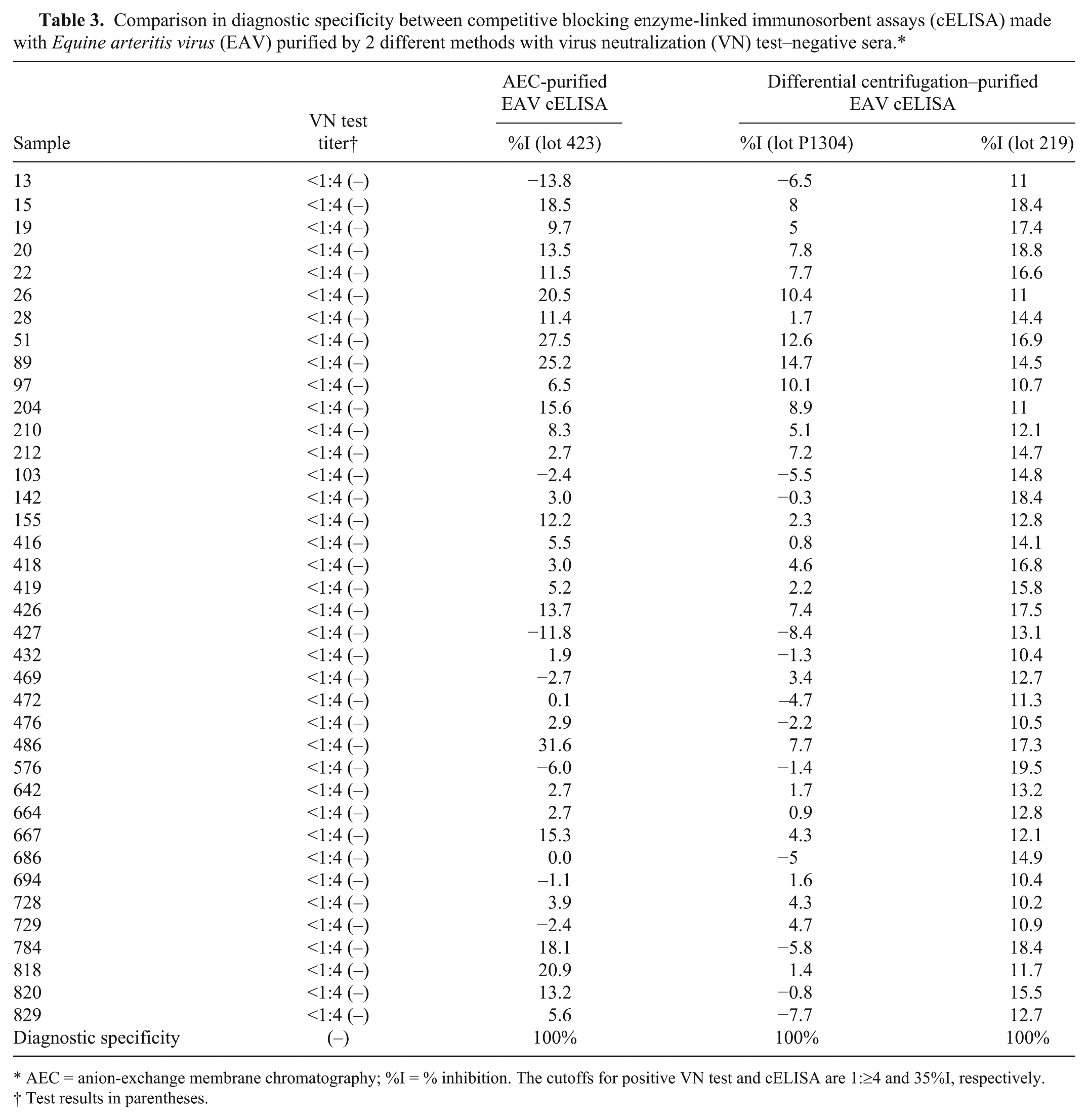
AEC = anion-exchange membrane chromatography; %I = % inhibition. The cutoffs for positive VN test and cELISA are 1:≥4 and 35%I, respectively.
Test results in parentheses.
Comparison of EAVs purified by 2 different methods using Western blots with GP5-specific mAb 17B7
Western blot analysis using GP5-specific mAb 17B7 revealed clearly different band patterns between AEC-purified and differentially centrifuged EAV (Fig. 5). The AEC-purified EAV had only 2 bands, representing a major GP5 monomer and a minor GP5-M heterodimer (12.8 GP5 to GP5-M ratio; Fig. 5). In contrast, immunoblots of differentially centrifuged EAV had multiple bands including GP5, GP5-M, and several larger bands, all detected with GP5-specific mAb (Fig. 5). The ratio of GP5 to GP5-M in the differentially centrifuged EAV was 2.7 while the ratio of GP5 to larger bands was 0.5 (Fig. 5). Thus, the proportions of GP5 monomer found were ~86.3% in the case of the AEC-purified EAV and <29.4% in the case of differentially centrifuged EAV. Interestingly, the total number of silver-stained bands (area density 13,690) in the AEC-purified EAV was also significantly lower than that in differentially centrifuged EAV (area density 140,528), although both EAV antigens were loaded with the same amount of total protein per lane before SDS-PAGE (Fig. 6). Samples of both EAV antigen preparations were boiled with SDS and 2-mercaptoethanol before separation by SDS-PAGE, which should have dissociated most noncovalent and disulfide bonds. Despite this, large molecules containing epitopes reacting with the GP5 mAb were detected in the Western blot. These large molecules may have been differentially processed GP5 precursors or other covalently linked molecules. Such differences in molecular forms containing GP5 epitopes noted in Western blots and the total number of silver-stained bands may be involved in exposure and reactivity of the epitopes to the mAb in the cELISA, resulting in increased sensitivity using the AEC-purified EAV.
Comparison between anion-exchange membrane chromatography (AEC)-purified and differential centrifugation–purified Equine arteritis virus (EAV) in Western blot using EAV glycoprotein 5 (GP5)-specific monoclonal antibody 17B7. Lanes A and B contain 1.77 μg and 0.885 μg of protein per lane of AEC-purified EAV, respectively; lanes C and D contain 1.77 μg and 0.885 μg of protein per lane of differential centrifugation–purified EAV, respectively.

Comparison between silver-stained bands of anion-exchange membrane chromatography (AEC)-purified and differential centrifugation–purified Equine arteritis virus (EAV). Lane A: 1.77 μg of protein per lane of AEC-purified EAV; lane B: 1.77 μg of protein per lane of differentially centrifuged EAV.
Discussion
Equine arteritis virus is an equine infectious disease that is known to occur on all continents.2,10,13,16,20,21 Each year, ~3,000 major equestrian events occur globally, requiring increasingly short quarantine times for entry to event-hosting countries and providing the potential risk of spread of EAV and other equine respiratory pathogens (Cooke et al., Facilitating safe international movement of horses for the purpose of participating in equestrian events, Proceedings of the 9th international conference on equine infectious diseases, October 2012, Lexington, Kentucky). Under such circumstances, rapid and accurate screening for EAV with respect to horses involved in national as well as international movement is integral to controlling the transmission of EAV. In 2013, a cELISA capable of detecting antibody to a nonneutralizing epitope in EAV GP5 was developed and shown to have an improved resolution and diagnostic performance7,8 when compared with a previously reported cELISA 6 and previously reported ELISAs.4,19 Based on field trials, the improved cELISA had a diagnostic sensitivity of 98.2% and a diagnostic specificity of 99.5%. 8 The evidence provided by our study would indicate that the cELISA can be considered a reliable alternative to the most sensitive EAV antibody assay, the VN test. Because the cELISA is easier to run, more robust, and more rapid, it could be useful in countries that face difficulties in being able to perform the OIE-prescribed VN test. However, the detection of some VN test–positive sera with low antibody titers that were negative by cELISA would suggest that there is room for even further improvement in harmonization between the VN test and the cELISA (Pfahl et al., Evaluation of a commercially available competitive ELISA). In our study, the sensitivity of the cELISA was improved relative to the OIE-prescribed VN test for detecting antibodies to EAV by using AEC-purified virus as the coating antigen in the assay.
Several sets of data in the present study demonstrated that AEC-based purification of EAV and its use in an antibody cELISA improved sensitivity and increased harmonization with results obtained with the OIE-prescribed VN test performed by the OIE EVA Reference Laboratory at the University of Kentucky (Lexington, Kentucky). First, the analytical sensitivity of the cELISA was improved using AEC-purified EAV as 2- to 4-fold higher dilutions of EAV positive sera were detected. Second, the AEC-purified EAV cELISA detected antibody 1 day earlier than was found using a differential centrifugation–purified EAV cELISA when testing sequential sera from an MLV-vaccinated horse. Third, there was significantly higher agreement (82.5%) between VN test results and AEC-purified EAV cELISA results based on testing 40 borderline VN test–positive samples than with the cELISA developed with differentially centrifuged EAV (32.5% agreement). Fourth, the higher analytical sensitivity of the cELISA developed with AEC-purified EAV was consistently repeatable in several runs by an operator in 1 laboratory, and reproducible between different operators in independent laboratories. Fifth, aside from improved sensitivity, diagnostic specificity of the AEC-purified EAV cELISA was similar to the cELISA developed with differentially centrifuged EAV when compared using 2 specificity check sets containing 38 sera with high negative %I (>10%, but <35%) and 45 sera with average negative %I (<10%).
The evaluation of EAV antigens purified by AEC and differential centrifugation by SDS-PAGE followed by silver staining and Western blotting using GP5-specific mAb 17B7 clearly demonstrated a difference in the relative purity and quality of the GP5 monomer containing cELISA target epitope. One possible reason for the efficiency of AEC purification of EAV GP5 is that it has a calculated isoelectric point of 4.99, whereas the isoelectric point of the overall structural proteins of EAV is 8.37. Therefore, at a pH of 7.2, which was used in AEC purification, GP5 would carry a net negative charge and, accordingly, bind strongly to the AEC capsule membrane. This could contribute to the amount and purity of the GP5 obtained in the final purification product. Furthermore, the EAV purified by differential centrifugation likely contained only GP5 associated with virions and possible cell membrane fragments. These differences in the GP5-containing end-product obtained by the 2 different purification procedures likely contributed to the improved cELISA sensitivity of the AEC-purified antigen. The AEC-based purification method was fast and efficient, and it removed 99.9% of the EAV added to the capsule (Table 4).
Summary of 2 Equine arteritis virus (EAV) purification methods.*
Starting virus volume and titer were 1.75 liters and 106.3 TCID50/mL, respectively. AEC = anion-exchange membrane chromatography; TCID50 = 50% tissue culture infective dose; cELISA = competitive blocking enzyme-linked immunosorbent assay.
An improved method of EAV purification was developed based on an easily scalable protocol using AEC. A cELISA using AEC-purified EAV as coating antigen had a 2- to 4-fold increase in analytical sensitivity and was 82.5% in higher agreement with the results using the VN test than that obtained with the cELISA based on EAV purified by differential centrifugation. The level of agreement in the results derived using the EAV antibody cELISA and the OIE-prescribed VN test, together with previously published data on this cELISA,7,8 provide further support for its use as an alternative to the OIE-prescribed VN test for the serodiagnosis of EAV.
Footnotes
Acknowledgements
We thank Ms. Kathleen M. Shuck for technical support and Mr. Walter Heiniger for animal care.
Authors’ contributions
TC McGuire contributed to conception of the study and critically revised the manuscript. CJ Chung contributed to conception and design of the study; contributed to acquisition, analysis, and interpretation of data; drafted the manuscript; and gave final approval. UBR Balasuriya contributed to design of the study. PJ Timoney contributed to acquisition, analysis, and interpretation of data. AL Grimm, CL Wilson, UBR Balasuriya, G Chung, and C-B Bandaranayaka-Mudiyanselage contributed to acquisition and analysis of data. SS Lee contributed to analysis of data. All authors agree to be accountable for all aspects of the work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
a.
Eagle minimal essential medium, American Type Culture Collection, Manassas, VA.
b.
Fetal bovine serum, PAA Laboratories, Piscataway, NJ.
c.
Triton X-100, Sigma-Aldrich Corp., St. Louis, MO.
d.
Sartobind Q Singlesep, Sartorious Corp., Bohemia, NY.
e.
ARVAC, Zoetis, Florham Park, NJ.
f.
High binding 96-well plates, Costar Stripwell (Costar 2592), Corning Inc., Vernon Hills, IL.
g.
ELISA blocking buffer, VMRD Inc., Pullman, WA.
h.
Mylar bags, IMPAK Co., Los Angeles, CA.
i.
ELISA antibody diluting buffer, VMRD Inc., Pullman, WA.
j.
Goat anti-mouse immunoglobulin G conjugated to horseradish peroxidase, VMRD Inc., Pullman, WA.
k.
TMB substrate solution, SurModics Inc., Eden Prairie, MN.
l.
Stop solution, SurModics Inc., Eden Prairie, MN.
m.
Multiskan MCC/340 ELISA reader, Titertek Instruments Inc., Huntsville, AL.
n.
Monoclonal antibody 17B7, VMRD Inc., Pullman, WA.
o.
SDS-PAGE sample buffer, DGel Sciences, Montreal, Quebec, Canada.
p.
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis gel, Bio-Rad Laboratories, Hercules, CA.
q.
Silver stain kit, Pierce Biotechnology Inc., Rockford, IL.
r.
Peroxidase-conjugated goat anti-mouse anti-bovine IgG, KPL Inc., Gaithersburg, MD.
Declaration of conflicting interests
The author(s) declare the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Authors Chungwon J. Chung, Amanda L. Grimm, Carey L. Wilson, Grace Chung, and Chandima-Bandara Bandaranayaka-Mudiyanselage are employed by VMRD Inc. The remaining authors declare no conflicting interests with respect to their authorship or the publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by VMRD Inc.