
Abstract
To investigate the prevalence and genetic characterization of Pigeon circovirus (PiCV) circulating in Chinese flocks, the genomic DNA of 144 samples collected from pigeons in 6 different geographic regions of eastern China between 2009 and 2010 were amplified using previously published PiCV primers. The PiCV sequence was detected in 83 of 104 unhealthy pigeons (79.8%) and 25 of 40 healthy pigeons (62.5%). The overall positive rate was 75% for all samples. An inverse primer polymerase chain reaction (IP-PCR) assay was performed to amplify the full-length sequence from a random sample of each region, and 6 specific DNA fragments were gel-purified and sequenced. The 6 full-length sequences were designated as SHWH-AB4 (2,031 bp), NJPK-21 (2,035 bp), HBLF-E2 (2,031 bp), JSJN (2,039 bp), SDDZ (2,037 bp), and AHBZ (2,035 bp) after BLAST analyses. The phylogenic tree and amino acid comparison indicated that all the strains examined were derived from a common strain, but had undergone genetic mutations through time. Pairwise comparisons revealed 93.4%–100% amino acid identity for the putative replication-associated proteins and 67.5%–100% for the putative capsid proteins.
Introduction
The first Pigeon circovirus (PiCV; family Circoviridae, genus Circovirus 11,24,26 ) infection was documented in Canada in 1986, 27 but has now spread to numerous countries. Infection is characterized by a broad range of clinical symptoms, including lethargy, anorexia, weight loss, respiratory distress, and diarrhea. 8,13,16,17,22 A comprehensive study demonstrated that PiCV can lead to young pigeon disease syndrome (YPDS), 14 which is a multifactorial disease that causes immunosuppression in infected pigeons. 12 YPDS has been commonly found in young racing pigeons for more than 2 decades. Infected pigeons displayed poor racing performance and high morbidity and mortality rates between the 3rd and 20th week after hatching. 4 YPDS is characterized with nonspecific clinical symptoms, such as anorexia, depression, ruffled feathers, vomiting, diarrhea, polyuria, and a fluid-filled crop. 4 Typical pathological findings induced by circoviral infection are atrophy of the bursa of Fabricius and thymus. 19 Many of the symptoms or signs are attributable to secondary infections, as a result of PiCV-associated immunesuppression. 1,14 These secondary infections are commonly caused by Chlamydophila (ornithosis/psittacosis), herpes virus, Pasteurella (cholera), Paramyxovirus-1, Trichomonas (canker), and Aspergillus, among others. 1 To date, YPDS and other conditions associated with the PiCV infections have become an economical threat to the pigeon industry worldwide.
Pigeon circovirus contains a circular, single-stranded DNA of approximately 2 kb in size within a nonenveloped protein coat. 23 The genome shares a low-level DNA homology with the psittacine Beak and feather disease virus (BFDV). 7,22 PiCV is currently classified as a tentative member of the family Circoviridae due to its genome similarity with the other members of the family. So far, the culture identification of PiCV has been unsuccessful. 3 However, electron microscopy and a method based on a specific DNA sequence demonstration can be used to detect the presence of the circovirus. 17,19
A 2009 report describes PiCV infections in the pigeon population in eastern China. 28 However, the genetic characterization of the PiCV strains in the region is not known. The objective of the present study was to characterize the prevalence and the genetic features of the PiCV strain that was infecting the pigeon population in eastern China.
Materials and methods
Sample sources
A total of 104 infected pigeons displaying clinical symptoms (such as respiratory signs, weight loss, and diarrhea) and 40 healthy pigeons were collected from 12 different regions in 6 provinces (Shandong, Anhui, Zhejiang, Shanghai, Fujian, and Jiangsu) in eastern China between 2009 and 2010 (Table 1). The 12 regions were chosen because they had the highest pigeon population density in China. The tissue samples were collected from the pigeons and immediately frozen at 80°C.
Molecular characterization and epidemiological investigation of Pigeon circovirus isolated in eastern China: sources and detection results.*
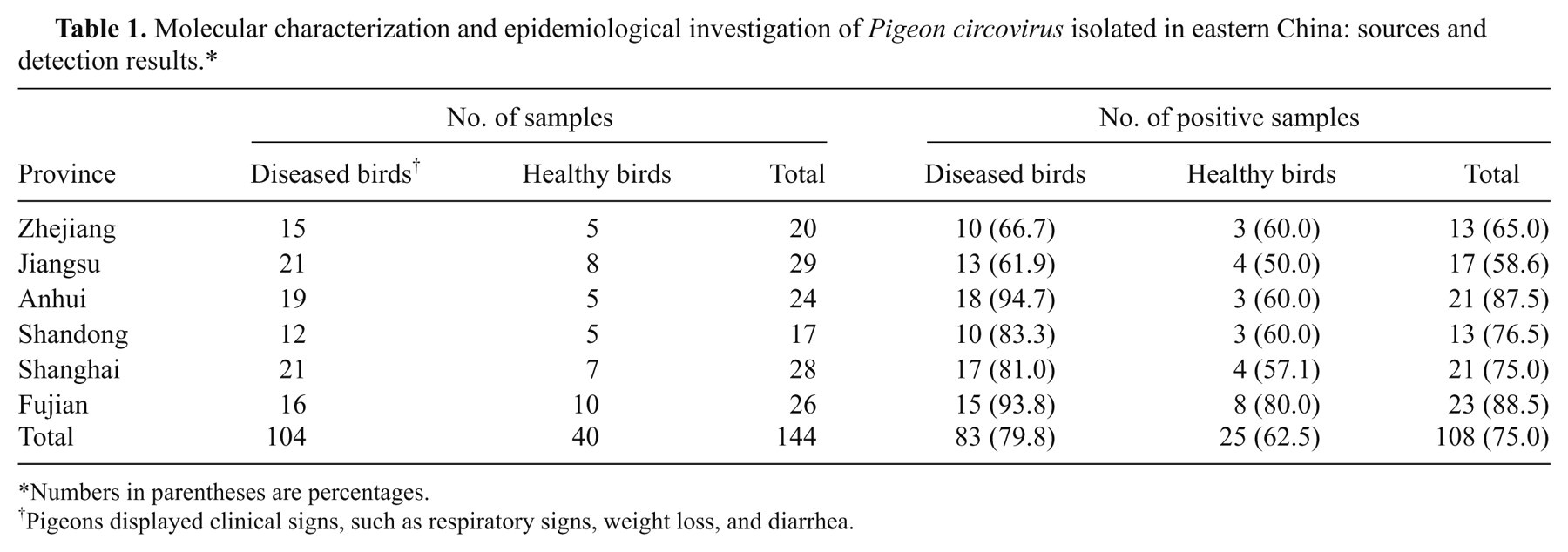
Numbers in parentheses are percentages.
Pigeons displayed clinical signs, such as respiratory signs, weight loss, and diarrhea.
Nucleic acid isolation
The genomic DNA was extracted from 200 µl of tissue homogenate (10%, w/v) or from approximately 25 mg of tissue samples using a commercial kit. a The extracted DNA was eluted twice with 25 µl of sterile distilled water to give a final extract volume of 50 µl.
The detection of PiCV by polymerase chain reaction
To detect the presence of PiCV sequence, the extracted genomic DNA was PCR amplified using PiCV-specific primers (forward: 5’-TTGAAAGGTTTTCAGCCTGGC-3’; reverse: 5’-AGGAGACGAAGGACACGCCTC-3’) as previously described. 4 The expected PCR product was 325 base pairs (bp) in size. The PCR mixture (50 µl) contained 40.5 µl of water, 5 µl of 10× buffer, 1 µl of each deoxyribonucleotide triphosphate (2.5 mM each), b 2 µl primers (10 µM each), 1 µl of template DNA (extracted as described above) and 0.5 µl of Taq DNA polymerase (5 U/µl). b The PCR reaction was conducted in a thermal cycler c using the following conditions: denature for 5 min at 94°C, followed by 40 cycles of 94°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec, and elongation for 5 min at 72°C. The amplicons were resolved by electrophoresis in a 2% agarose gel, which was stained with ethidium bromide (0.5 g/ml) in Tris–borate–ethylenediamine tetra-acetic acid buffer (89 mM Tris; 89 mM boric acid; 2 mM ethylenediamine tetra-acetic acid, pH 8.4) and visualized under ultraviolet light.
Inverse primer polymerase chain reaction on the PiCV genome
A positive DNA sample extracted above from each region were amplified with inverse primer polymerase chain reaction (IP-PCR) using the inverse primer pair 5’-CCCCACTGAAGAAGAGATTAAGAGCCTGGAAA CGTG-3’ (forward) and 5’-CTTAATCTCTTCTTCAGTGGGGTT GTTCAA-3’ (reverse). Six full-length clones were obtained in the current study, and each PCR product was approximately 2,000 bp in size. The IP-PCR reagents were the same as described above, and the amplification conditions were set as following: 30 cycles of 94°C for 45 sec, 53°C for 1 min, and 72°C for 2 min with an initial denaturation of the template DNA at 94°C for 5 min. The PCR products with the expected sizes were purified and sequenced after subcloning into the pMD18-T vector with a Taq-amplified cloning kit according to the manufacturer’s protocol. b
The phylogeny of the PiCV strains
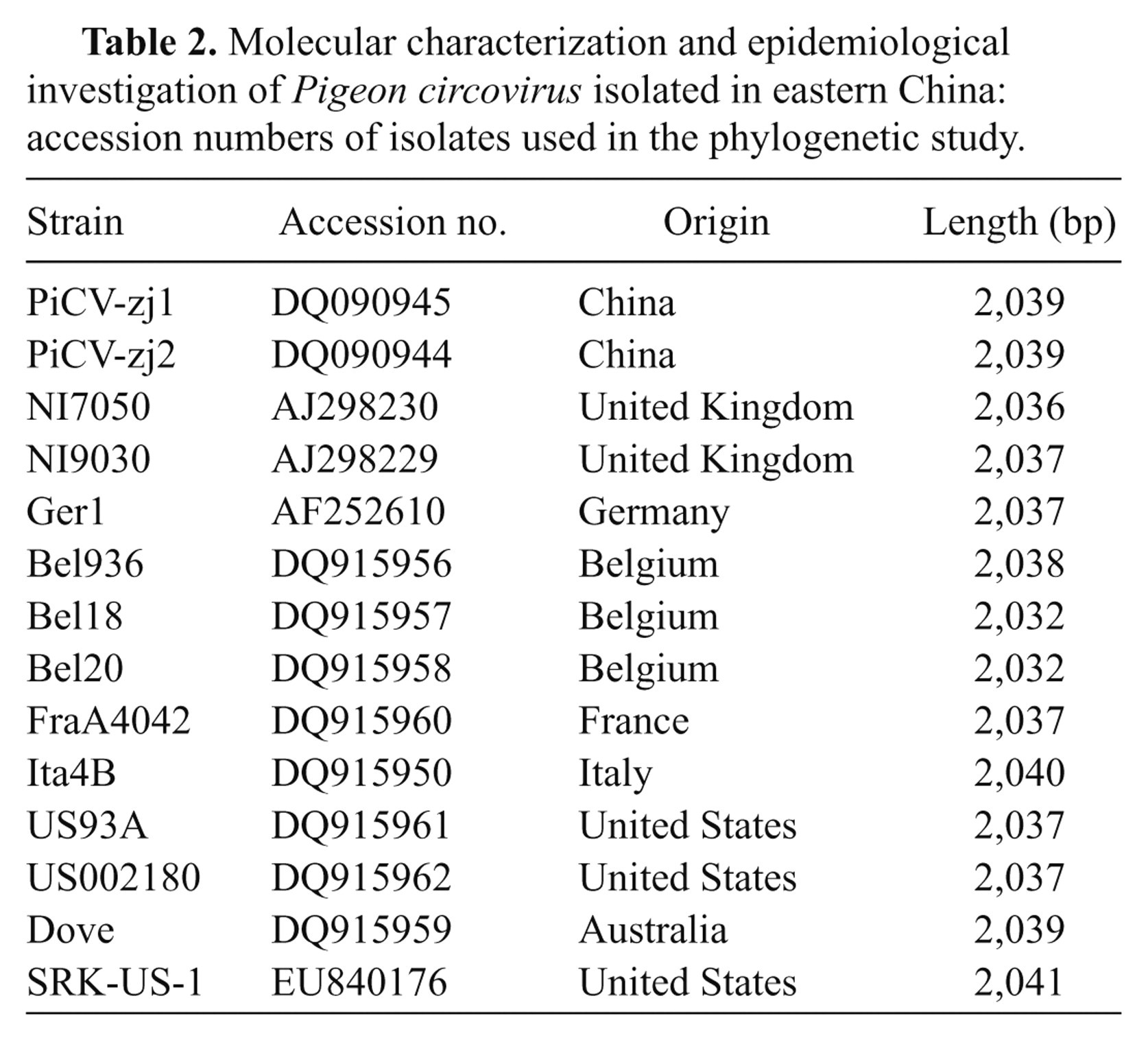
The DNA sequences of the 6 PiCV strains obtained in the current study were compared with 14 published full-length PiCV sequences from the GenBank database (Table 2) using the sequence analysis software DNASTAR d and ClustalW. 6,20 The phylogenetic tree was generated using the distance-based, neighbor-joining program MegAlign. 6,10 The bootstrap values were calculated on 1,000 replicates of the alignment.
Molecular characterization and epidemiological investigation of Pigeon circovirus isolated in eastern China: accession numbers of isolates used in the phylogenetic study.
The amino acid sequence analysis of the replication association and capsid proteins
The amino acidic sequences of the replication-associated (Rep) and capsid (Cap) proteins were aligned and compared. Sequence identity was analyzed using the DNASTAR d and ClustalW method. 6,20 By aligning the 6 PiCV sequences obtained in the present study using the algorithm ClustalW method included in the program MegAlign, the amino acids predicted from the open reading frame (ORF)2 nucleotide sequences were compared. 6,9
Results
Polymerase chain reaction testing of the clinical samples
A specific DNA fragment was PCR-amplified to produce the amplicons with expected size. The amplicons were then purified, sequenced, and further confirmed by BLAST (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi). A total of 108 samples out of 144 tissue field samples were positive for PiCV (Fig. 1). Among the PiCV-positive samples, 83 were detected in 104 diseased pigeon samples (79.8%), and 25 were detected in 40 healthy pigeon samples (62.5%).
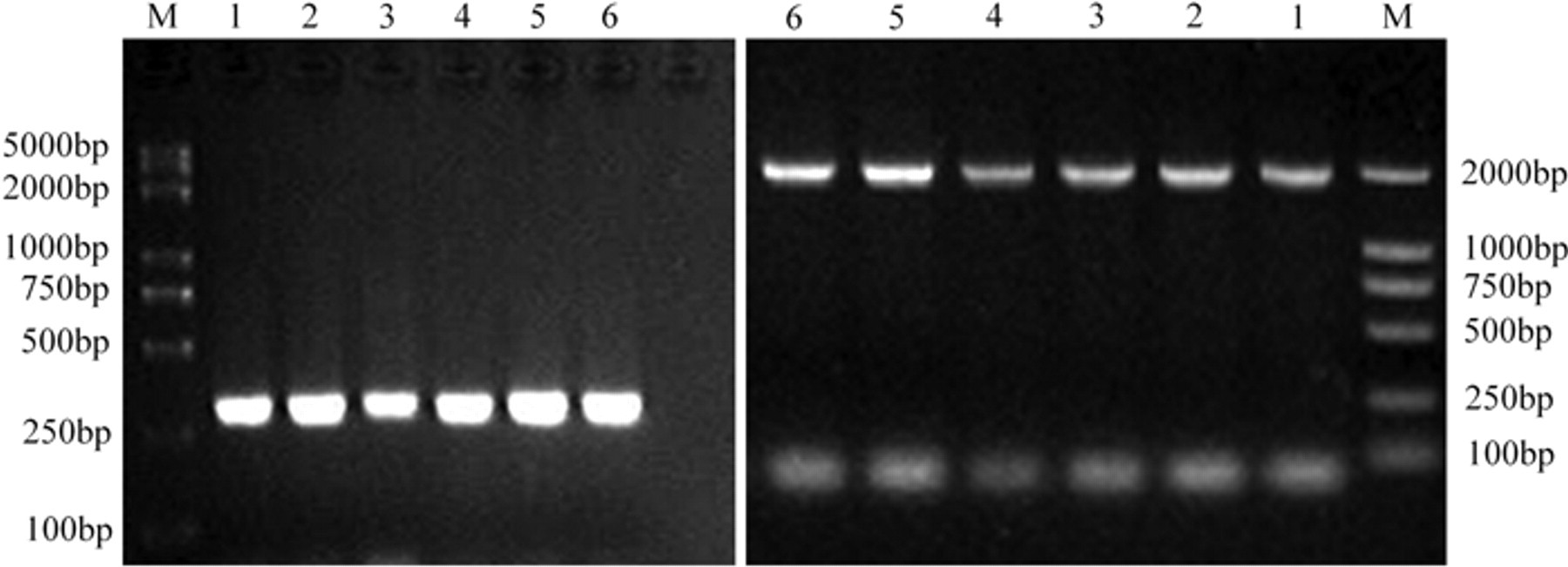
The result of Pigeon circovirus (PiCV)-specific DNA by polymerase chain reaction (PCR). Left panel: identification of PiCV by PCR amplification; right panel: amplification products of PiCV genome. Lane M: DNA marker; lanes 1–6: SHWH-AB4, NJPK-21, HBLF-E2, JSJN, SDDZ, and AHBZ; lane 7: negative control.
Inverse amplification of the PiCV genome
The IP-PCR technique was performed using PiCV-positive samples to produce 6 genomic DNA fragments (Fig. 1), which were gel-purified and sequenced. The sequences were confirmed by BLAST and designated as SHWH-AB4 (2,031 bp), NJPK-21 (2,035 bp), HBLF-E2 (2,031 bp), JSJN (2,039 bp), SDDZ (2,037 bp), and AHBZ (2,035 bp).
The phylogeny of the PiCV
A phylogenic tree was constructed using the MEGALIGN software program d for the 6 PiCV genomic sequences identified in the present study and the 14 additional PiCV sequences published in the GenBank database (Fig. 2). The PiCV phylogenic tree was divided into 2 branches. All 6 strains detected in the current study belonged to the same branch. The Dove strains that were isolated from Australia solely constituted the second branch. In the dendrogram, the sequences of the 3 isolates (DQ090945 PiCV-zj1 China, JSNJ China Seq, and DQ090944 PiCV-zj2 China) formed a cluster and collectively differed from the remaining 5 isolates.
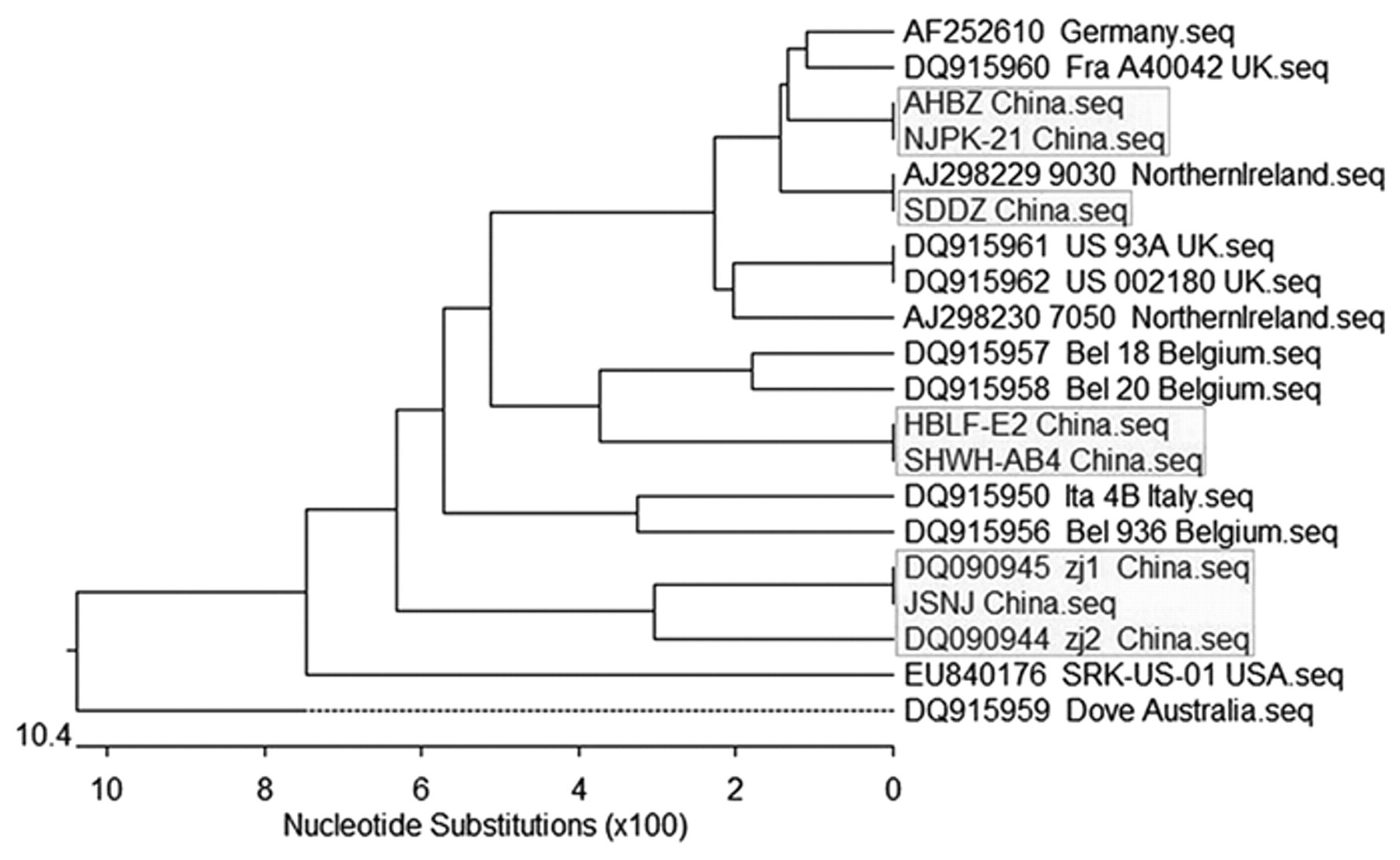
Phylogenetic tree based on the comparison of entire Pigeon circovirus (PiCV) genomes. The PiCV isolates originating from China are marked in bold. The phylogenetic tree was constructed using the MegAlign program.
The amino acid sequence analysis of the Rep and Cap proteins
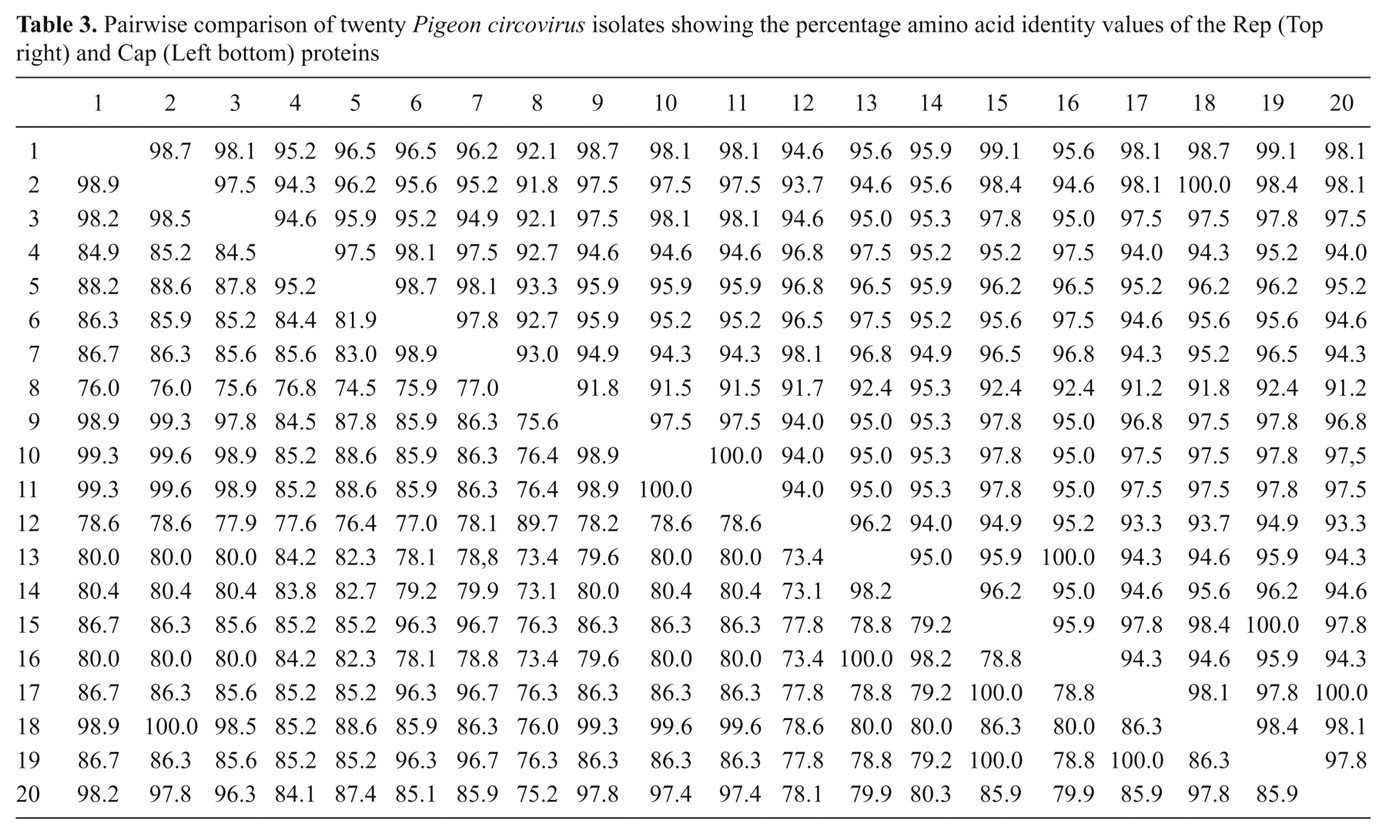
The amino acid sequences were translated from the sequenced PiCV genome using the ClustalW program. The Rep and Cap protein homologies among the isolated strains in the present study ranged from 93.4%–100% and 67.5%–100%, respectively. They also ranged from 90.3%–100% and 68.2%–100%, respectively, among the published full-length PiCV sequences from the GenBank database (Table 3).
Pairwise comparison of twenty Pigeon circovirus isolates showing the percentage amino acid identity values of the Rep (Top right) and Cap (Left bottom) proteins
The Rep protein present in all 20 PiCV strains consisted of either 315 or 317 amino acids (aa). The difference in size was due to a 2 aa deletion at residues 3 and 4 in the US93A, Bel18, Bel20, Bel936, and Ita4B strains (Fig. 3). All of the strains collected in eastern China in the current study encoded the 317 aa Rep protein. It was also found that the Rep protein from 12 PiCV strains contained motifs associated with rolling circle replication (RCR), namely FTLNN, T–LQGF, CSK, and G-GKS, which are putatively associated with dNTPase activity. These motifs were fully conserved in all 20 PiCVs. 23

Alignment of the full-length amino acid sequences of Rep among 20 strains. Amino acid deletions are indicated by shaded. Conserved sequences and the rolling circle replication motifs are boxed with solid lines.
The size of the Cap protein found in the strains varied greatly. Difference sizes included 270, 272, 273, or 274 aa, due to a single aa deletions at residues 7, 53 , 130, 180, 250, and 262 and double aa deletions at residues 28 and 29 (Fig. 4). A single deletion at residue 250 was exclusively found in the AHBZ strain. The N-terminal region of the Cap was highly basic and arginine-rich, which was commonly observed in other circoviruses. 18 For example, 25 of the first 50 aa were arginine in the AHBZ PiCV. The alignment data also identified 9 small variable regions 4–8 aa long. Among these variable regions, the most diverse ones were encompassing aa residues 34–36, 52–55, 103–108, 127–130, 188–195, and 214–223.

Alignment of the full-length amino acid sequences of Cap among 20 strains. Amino acid deletions are indicated by shading. The 6 variable regions with a relatively high number of aa substitutions are boxed with solid lines.
Discussion
Pigeon circovirus infections affecting the pigeons have been reported in the United States, Canada, China, Australia, 27 and a number of European countries. 2,5,15,17 In the current study, the genomic sequences of the PiCV strains obtained from the infected pigeons in 6 geographic regions of eastern China were identified. The PiCV infections were detected in 83 of the 104 diseased pigeons (79.8%) and in 25 out of the 40 healthy pigeons (62.5%). The overall positive rate was 75%, indicating that the PiCV-positive rate was very high in the pigeon populations in eastern China. Like other circoviruses, PiCV is thought to suppress the host’s immune system and thus increase the likelihood of secondary infections. 21
The genetic diversity of the virus may lead to variations in their pathogenicity. Porcine circovirus−1 and −2 (PCV-1 and PCV-2) are both small viruses composed of a single-stranded circular DNA. While PCV-1 is a nonpathogenic virus that is commonly found in domestic swine, PCV-2 is a pathogenic virus that shares 76% sequence identity and similar genomic organization with PCV-1. The pathogenicity of PiCVs detected in the current study and their relationship with PCV-1 and PCV-2 need to be further examined.
The major advantage of IP-PCR is the ability to amplify an unknown flanking region using 2 specific primers, which could avoid sequence assembly and minimize interference from other circovirus-like agents. A 2001 study found that the positive rate of PCV-2 detection using PCR was enhanced by the amplification of PCV-2 ORF2 rather than the amplification of PCV-2 ORF1 or the complete PCV-2 genome. 24 In the same study, the authors suspected that contaminants having similar sequences to the PCV-2 ORF2 were present in porcine sera and, therefore, may have led to PCR interference. 25
Pairwise comparisons showed that the genome sequence of PiCV detected from samples isolated from pigeons in eastern China appeared to be closely related. The sequence identity was found to be 98.9%–100%, and the genome size ranged from 2,031 nt (SHWH-AB4 and HBLF-E2) to 2,039 nt (JSJN, PiCV-zj1, and PiCV-zj2). The PiCV-zj1, PiCV-zj2, and NJPK strains were isolated from the Zhejiang Province, while the JSNJ strains were isolated from Jiangsu Province. However, the PiCV-zj1, PiCV-zj2 strains (2005) were found closely related to the JSNJ strains (2009), but not from the NJPK strains (2009). Results indicated that geographical and time factors had almost no effects on the genome diversity of PiCV.
The phylogenic tree and aa sequence comparison found that the epidemic PiCV strains in eastern China had low genomic separation and diversity. The variation was mainly due to the variability within the Cap proteins (78.8%–98.2% identity), while the Rep proteins were highly conserved (94.30%–100%) in all strains. The PiCV genome contained a large number of base insertions, deletions, and other mutations. The phylogenic tree indicated that the SDDZ strain isolated in eastern China originated from the AJ2982299030 strain isolated in Northern Ireland. This link between the 2 strains may be caused by the introduction of Northern Ireland breeding pigeons in Shandong Province in recent years. The PiCV-carrying healthy pigeons introduced from abroad could most likely be the origin of the virus in Chinese pigeons. Whether potential mutations in these carriers can suppress immunosuppression or via other means reduce the pathogenicity of PiCV require further studies.
Footnotes
a.
QIAamp® DNA Mini Kit, Qiagen Ltd., West Sussex, United Kingdom.
b.
Takara Bio Inc., Otsu, Shiga, Japan.
c.
Eppendorf Canada, Mississauga, Ontario, Canada.
d.
DNASTAR Inc., Madison, WI.
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The current study was supported by the Program for New Century Excellent Talents (NCET) in the University (no. NCET-08-0794) of China, the Qing Lan Project, China, Natural Science Foundation of Jiangsu Province (BK2008059), Research Foundation of Jinling Institute of Technology (no. Jit-n-2009-011), Natural Science Foundation of the Higher Education Institutions of Jiangsu Province, China (no. 10kjd230003), and Graduate Research and Innovation Project of Jiangsu Province (CX09B_ 246Z).