
Abstract
Mucopolysaccharidosis type IIID is caused by a deficiency of N-acetylglucosamine-6-sulfatase, which is one of the enzymes involved in the catabolism of heparin sulfate. Simple molecular marker assays underpin modern routine animal breeding and research activities worldwide. With the rapid growth of single nucleotide polymorphism (SNP) resources for many important animal genetic disorders, the availability of routine assays for genotyping SNPs is of increased importance. In the current study, real-time polymerase chain reaction (PCR) is demonstrated to provide a valuable approach as a rapid and accurate alternative to a previously developed gel-based PCR as a straightforward and efficient assay for the diagnosis of caprine mucopolysaccharidosis IIID.
The mucopolysaccharidosis type IIID (MPS IIID) disorders are lysosomal storage diseases. This genetic deficiency is characterized by deficient lysosomal hydrolase activity and subsequent lysosomal accumulation of uncatabolized heparin sulfate (HS) glycosaminoglycans (GAGs) material, which is normally degraded by the LH enzyme. 9,15 In caprine MPS IIID, the deficiency is mainly in the N-acetylglucosamine-6-sulfatase (G6S) activity with a perturbed catabolism of HS. This primary accumulation of HS is associated with a secondary accumulation of gangliosides and alteration of many lysosomal enzyme activities. 10,11
Mucopolysaccharidosis was first identified in humans, where it is known as Sanfilippo syndrome. 2 The disease is characterized by severe, progressive mental deterioration and increased urinary excretion of partially degraded HS but rather mild visceral and skeletal abnormalities. 1,18 Animal models for this syndrome include feline, canine, and caprine examples, where the disease occurs naturally. 6,7 In caprine MPS IIID, the G6S deficiency is associated with a single mutation. 4 The consequent lack of G6S activity in goats leads to the primary accumulation of uncatabolized HS-GAGs in lysosomes and marked cytoplasmic vacuolation in the central nervous system and somatic tissues. 14 Clinical signs exhibited by affected goats included delayed motor development, growth retardation, and early death.
Mucopolysaccharidosis IIID in goats has been described and characterized. 10 It appears to be confined to Nubian goats and their crosses; other breeds seem not to have this particular genetic mutation. 19 The predicted genotypic frequency for this disorder has been reported to be approximately 74.2%, 23.9%, and 1.9% for normal, carrier, and affected Nubian goats, respectively. 8 The molecular base for this disorder is a nonsense mutation, changing a C to T in codon 102 of the 559-amino acid G6S protein. 3 A goat that has 2 copies of the normal G6S gene is considered “normal.” The defective G6S mutation is a simple recessive gene, which means that a goat that has only 1 copy of it will appear normal and will not show any clinical signs. Such a goat is referred to as a carrier. The offspring that inherits the defective gene from both parents shows clinical signs and is referred to as affected.
The disease can be accurately diagnosed only by the identification of the point mutation by genetic analysis. Recently, a gel-based polymerase chain reaction (PCR) assay and a subsequent AluI restriction enzyme digestion was shown to differentiate the genetic forms of this allele combination. 13 Similarly, the same approach has been used for the identification of Sanfilippo syndrome in humans. 1
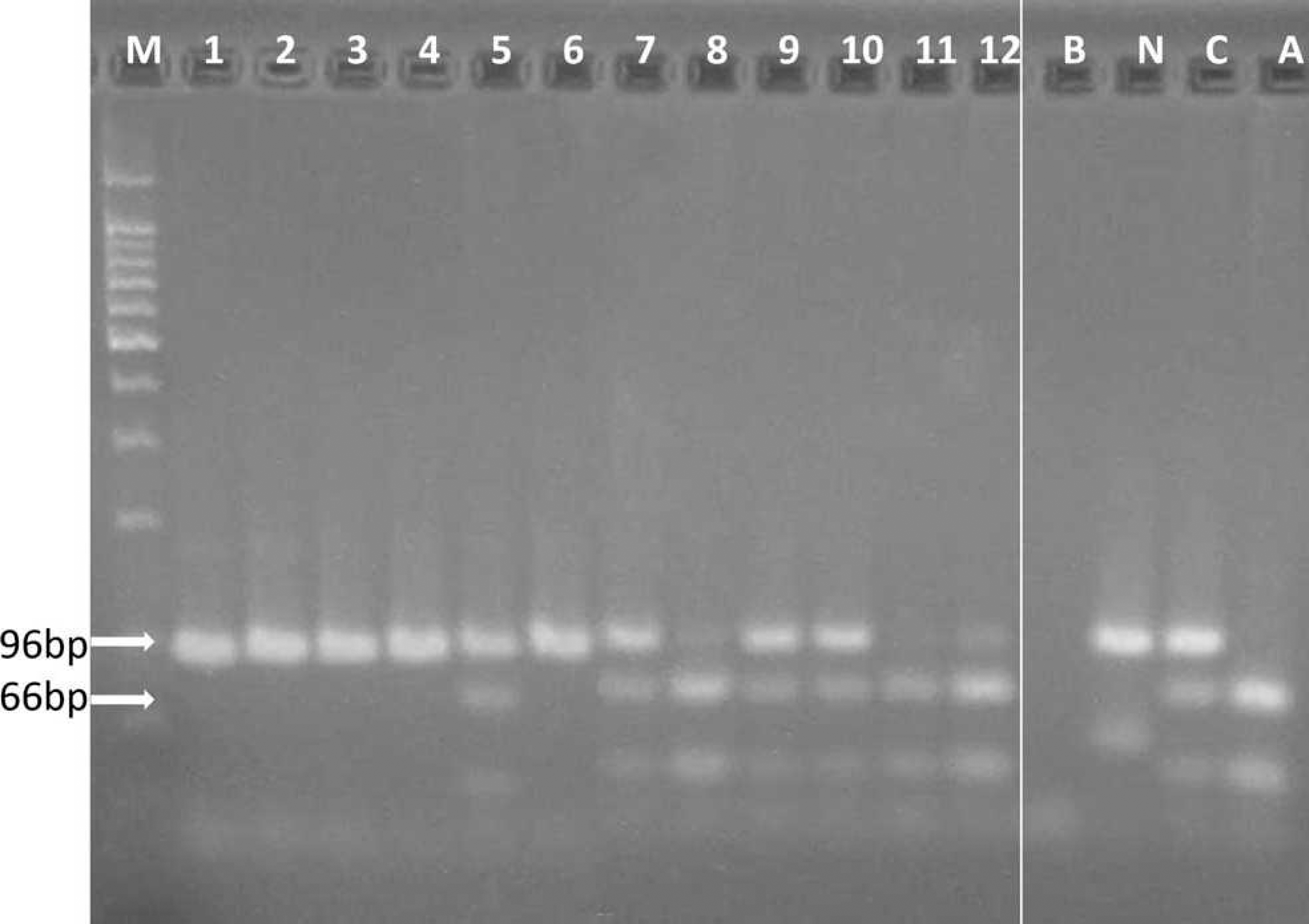
Gel-based polymerase chain reaction (PCR) for the identification of caprine mucopolysaccharidosis type IIID. After DNA amplification, a 96-bp DNA fragment containing the N-acetylglucosamine-6-sulfatase mutation site was produced. This amplicon was then digested using AluI restriction endonuclease. Cleavage of PCR products to 66- and 30-bp fragments indicated an affected animal (lanes 8, 11, and 12). Normal animals had only the 96-bp band (lanes 1–4 and 6), and carrier animals had 96-bp, 66-bp, and 30-bp fragments (lanes 5, 7, 9, and 10). A nontemplate (B), normal (N), carrier (C), and affected (A) controls were run in all reactions.
In the present study, a real-time PCR single nucleotide polymorphism (SNP) assay was developed to facilitate accurate diagnosis of MPS IIID. For DNA analysis by both conventional and real-time PCR, genomic DNA was extracted from white blood cells by using 50 μl of whole blood collected in ethylenediamine tetra-acetic acid (EDTA) tubes. Blood was transferred into 1.5-ml micro-centrifuge tubes containing 500 μl of refrigerated red blood cell lysis buffer (155 mM NH4Cl, 10 mM NaHCO3, and 0.1 mM EDTA, pH 7.4); tubes were vortexed vigorously and placed at −20°C for 10 min. Samples were centrifuged for 1 min at 21,000 × g to pellet the cells, and the supernatant was gently pipetted and discarded. The pellet of white blood cells was resuspended by adding 100 μl of 50 mM NaOH. Finally, the sample was boiled at 100°C for 10 min. After cooling the sample on ice, 5 μl of 2 M Tris- HCI, pH 8.0, was added to neutralize the sample pH. The genomic DNA obtained in this way was diluted 10-fold with distilled H2O and used directly for PCR. The gel-based PCR and restriction fragment length polymorphism assay were performed as previously described. 13 Briefly, the forward (5′-CTTATGTGCCAAGTGCTCTC-3′) and reverse (5′-CCTCCAGAGTGTTGTTAACC-3′) primers were obtained from a commercial source. a The gel-based PCR produced a 96-bp PCR product by using 2.0 μl of target DNA in a 25-μl PCR reaction containing 2.0 mM MgCl2, 20 mM of each deoxyribonucleotide triphosphate, 2.5 U of DNA polymerase b in 1X reaction buffer, and 0.15 μg/μl of each primer oligonucleotide. The PCR reaction controls were included in all tests. Blood samples of confirmed normal, heterozygous (carriers), and affected goats to the G6S allelic combination were used as positive controls. c The amplification was carried out in a commercial automated thermal cycler. d The PCR parameters were 94°C for 10 min, following by 35 cycles of 94°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec. Final extension was 10 min at 72°C. Subsequently, 19 μl of PCR products and 1 μl of AluI (10 U/μl) c were incubated at 37°C overnight. The digested PCR products were analyzed on 3.0% agarose gel and were stained using a florescent nucleic acid gel stain. e The DNA bands (96 bp, 66 bp, and 30 bp) were visualized by ultraviolet transillumination (Fig. 1).
For the real-time PCR assay, a TaqMan assay-by-design SNP genotyping technique was used. f The sequences for the development of the allelic discrimination assays were derived from the normal gene of Capra hircus G6S messenger RNA (GenBank U17694.1). Minor groove binder (MGB) probes were used for both the normal (5′-VIC-CTGGCTCGGCTCGG-MGBNFQ-3′) and mutational (5′-6FAM-CTGGCTCAGCTCGG-MGBNFQ-3′) allele of G6S. The MGB probe forms extremely stable duplexes with a single-stranded DNA target, allowing for a shorter and more target-specific probe to be used for hybridization, which makes it an ideal candidate for the detection of SNPs. 5,16 The real-time PCR G6S assay uses 1 VIC f and 1 fluorophore 6-carboxyfluorescein (FAM) dye-labeled probes (Fig. 2) and 2 target-specific primers. The quencher molecule quenches the fluorescence emitted by the fluorophore when excited by the cycler's light source. As long as the fluorophore and the quencher are in proximity, quenching inhibits any fluorescent signals. When the probe is in solution, the 3-dimensional conformation of the probe brings the quencher and MGB in close proximity of the fluorescent label, and the fluorescence is quenched, inhibiting all fluorescent signals. When the probe anneals to a target sequence, the probe unfolds, moving the quencher away from the fluorescent label with the MGB entering the minor grove, allowing a strong fluorescent signal 12 (Fig. 2).
The forward (5′-GCTTATGTGCCAAGTGCTCTCT-3′) and reverse (5′-TGGGTACTTCCCTGTCAGGAT-3′) primers were synthesized commercially e and correspond to base positions 290–385 bp of the Capra hircus G6S messenger RNA. The amplification mixture contained 1X TaqMan Universal PCR Master Mix, e 900 nM of each forward and reverse primer, 250 nM each of the probes, 2.0 μl of target DNA in a total 25-μl PCR reaction. One non-template and 1 positive DNA control for each allele (normal, carrier, and affected) were included in the test. Amplification and fluorescence detection were conducted using a commercial sequence detector f with a program consisting of an initial step at 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 60 sec.
Using representative genomic DNA samples for each of the genotypic combinations of this allele in the real-time PCR assay, the probes can clearly differentiate between normal, homozygous (affected), and heterozygous (carrier) forms of the genetic mutation (Fig. 3). In a normal animal, only the VIC dye-labeled probe will react with the target. Alternatively, if the FAM dye-labeled probe reacts only with the target DNA, it indicates that the nature of this gene in the animal is homozygous and the animal will then be classified as affected for this genetic anomaly. When both probes (VIC and FAM) react with the target, both normal and mutational alleles are present, and the animal will be classified as carrier.
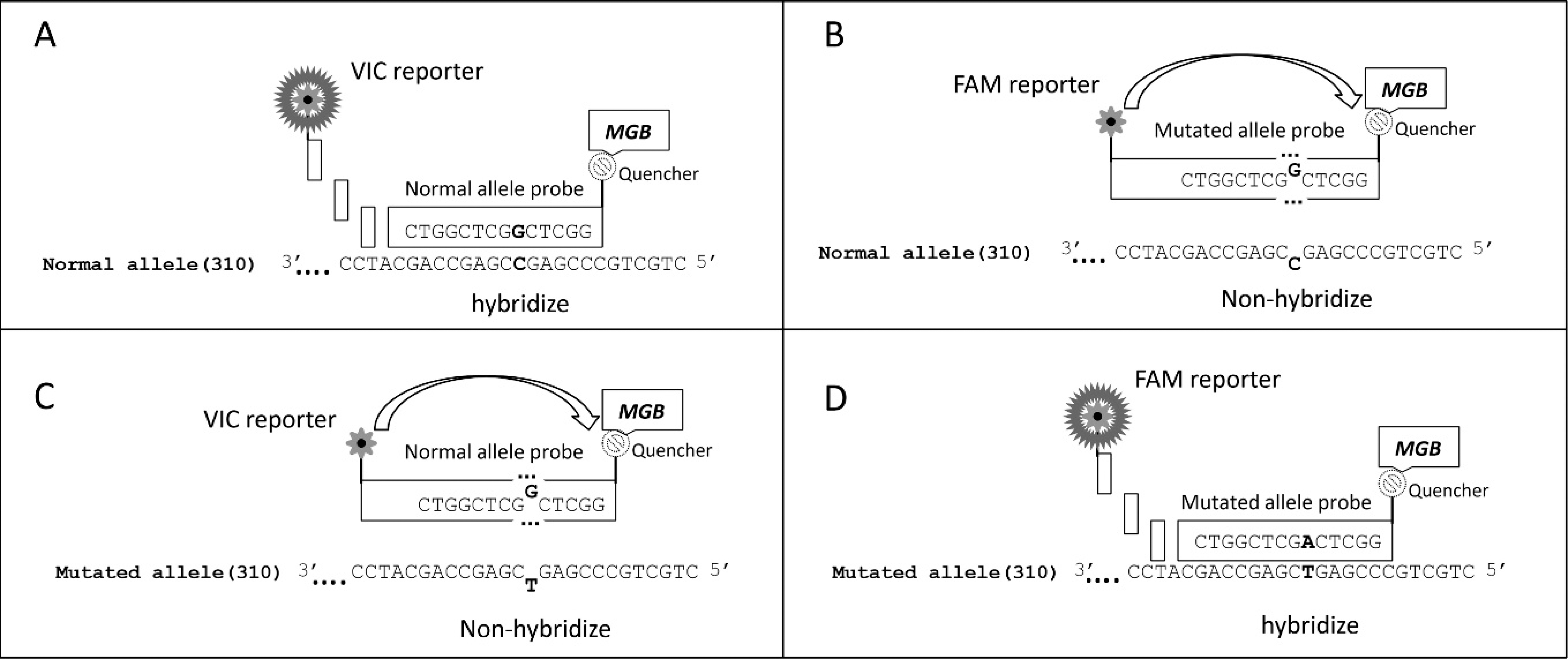
Real-time polymerase chain reaction allelic discrimination of the N-acetylglucosamine-6-sulfatase single nucleotide polymorphism (SNP). The SNPs affect fluorescent probe binding to complementary DNA, allowing discrimination of 2 alleles. Probes that are a perfect complement to the target DNA hybridize and are digested by the 5′→3′ exonuclease activity of Taq polymerase, separating VIC f or fluorophore 6-carboxyfluorescein (FAM) reporter from the quencher. Single base mismatched probes do not bind at this relative high annealing temperature (nonhybridize), remain intact, and do not report due to quenching of FAM or VIC fluorescence. MGB = minor groove binder probe.
For the evaluation of the real-time PCR assay and its comparative performance relative to the gel-based PCR, 100 EDTA blood samples from purebred Nubian goats, submitted to the diagnostic laboratory during a period of approximately 17 months, were tested. Results of the real-time assay indicated that 76%, 21%, and 3% of the goats were normal, carrier (heterozygous), and affected (homozygous), respectively. Results of the gel-based and real-time PCR tests reveal agreement between both assays for all samples, with the exception of 2 samples that were classified as affected by the real-time assay and inconclusive in the gel-based assay due to faint bands at 96 bp in addition to the expected 66- and 30-bp bands (Fig. 1, lanes 8 and 12).
To compare the overall performance of these 2 assays, historical data of 674 samples tested by the gel-based PCR from year 2000 to 2003 and 691 samples tested by real-time PCR from year 2004 to 2009 were evaluated (Table. 1). The Hardy-Weinberg model was used 17 to compare the results using these 2 populations. This model describes and predicts the genotype and allele frequencies in a nonevolving population, considering that a population's genotype and allele frequencies will remain unchanged over successive generations. 17 The calculated chi-square using this model illustrates the differences between expected and observed values for genotype counts (Table. 1). Results from both populations tested indicate that there is a 95% probability that the difference between observed and expected genotypes was caused by chance alone. The null hypothesis, that the observed and expected values are not significantly different, must be accepted. The populations are indeed in Hardy-Weinberg equilibrium. In other words, one can expect the allele frequencies to remain constant over time. Thus, the genetic structure of the population at this particular locus will remain in equilibrium as long as mutations remain negligible and there is not gene flow between populations by controlled mating. Results also indicate that in the goat population tested by the gel-based assay (year 2000–2003), the difference between observed and expected genotype is greater than in the population tested by the real-time PCR assay (year 2004–2009). This can be attributed to the reduced detection of affected goats by the gel-based assay with a +67% difference between observed and expected in the affected genotype compared with a +0.6% in the group tested by the real-time PCR assay. The limited difference between observed and expected normal and carrier genotypes is also considerable (+0.015% and −0.7%, respectively) in the population tested by the real-time PCR assay compared with the results obtained by the gel-based assay (+0.64% and −5%). The real-time PCR assay may be a better tool to accurately detect this genetic disorder and facilitate current efforts to control this mutation. Considering that this population is in Hardy-Weinberg equilibrium and that the epidemiological distribution of MPS IIID has been maintained unchanged, results indicate that the rate of diagnosis has persisted without considerable change during a 9-year period. Overall, 74–78% were diagnosed normal by both assays and 20–23% were carriers of this genetic marker. The real-time PCR assay unequivocally identified all samples, whereas the gel-based PCR assay yielded inconclusive results in 2 of 7 cases from affected goats (28%; Table 1, top).
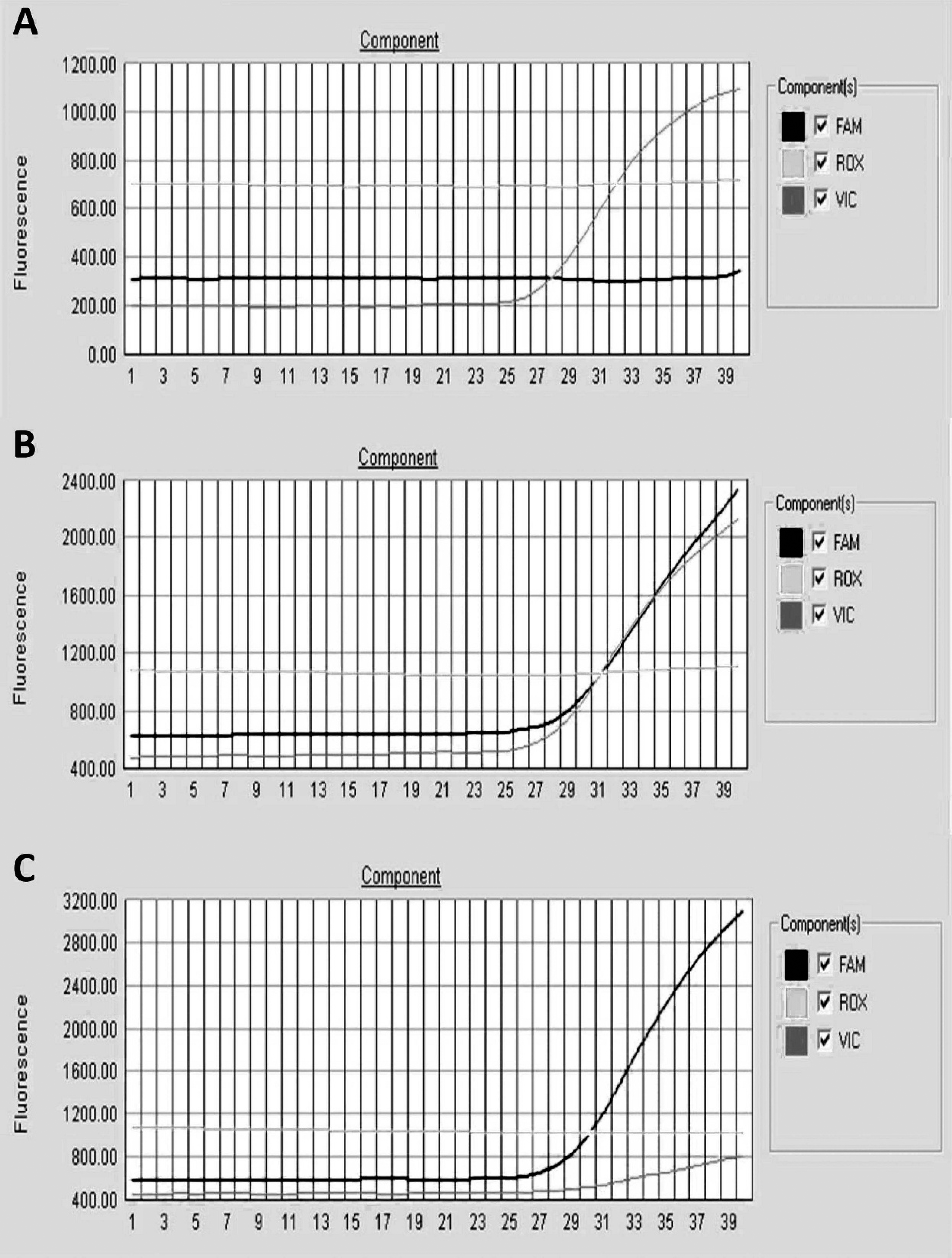
Real-time polymerase chain reaction (PCR) detection of caprine mucopolysaccharidosis type IIID using 2 minor groove binder probes that are able to differentiate a single point mutation.
Comparative allelic distribution of mucopolysaccharidosis IIID in the N-acetylglucosamine-6-sulfatase gene of goats tested by gel-based polymerase chain reaction (PCR) and real-time PCR, and observed and expected genotype distribution using the Hardy-Weinberg equilibrium model.*
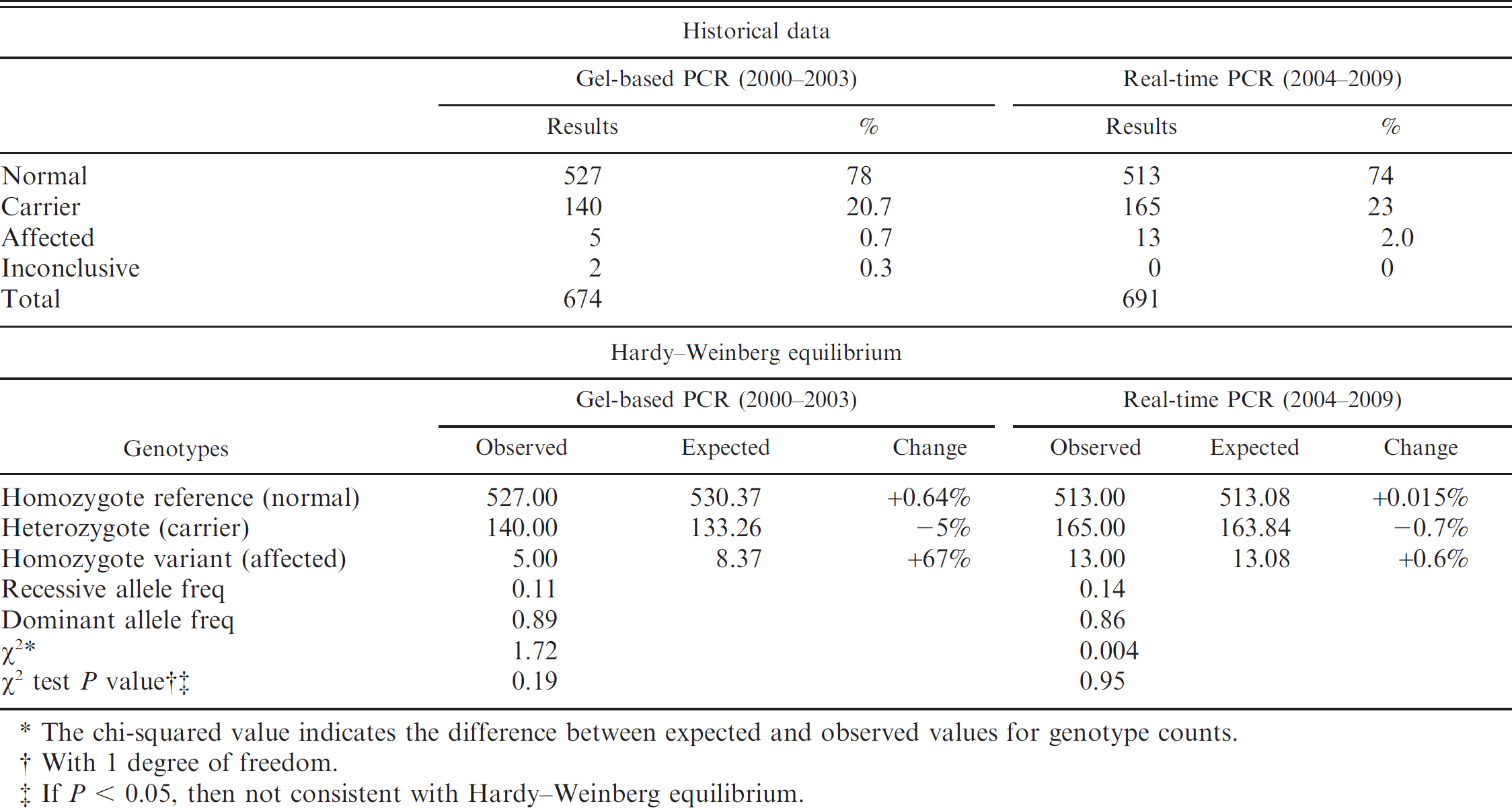
The chi-squared value indicates the difference between expected and observed values for genotype counts.
With 1 degree of freedom.
If P < 0.05, then not consistent with Hardy-Weinberg equilibrium.
Interestingly, the number of reported affected animals increased by 1% when using the real-time PCR assay. This may be attributed to the lack of inconclusive results reported in the real-time PCR assay compared with the inconclusive results frequently seen in the gel-based assay. In all cases, the inconclusive results were identified as affected by the real-time PCR assay (2/7 animals, 28%). Similar results were reported previously, when 552 Nubian goats were tested by gel-based PCR, where 73.5%, 25.2%, and 1.3% were classified as normal, heterozygous, and homozygous (affected) for the G6S mutation, respectively. 8
The real-time PCR assay reported in the present study for the diagnosis of caprine MPS IIID (G6S) is a robust and convenient genotyping assay. The assay is a single-step test and can replace the conventional gel-based PCR assay and restriction endonuclease digestion. The benefits of a real-time format for an accurate SNP genotyping using allele-specific probes may include increased specificity accuracy and reduced turnaround time compared with the gel-based PCR. The assay can also be incorporated into a high-throughput format with improved assay automation to enhance the genetics in Nubian goat herds across the country.
Acknowledgements
The authors thank the animal owners for the opportunity to use their samples for this test development; Dr. Heidi M. Hoard, Department of Pathology, Michigan State University, for providing initial positive control samples; and Sandra Rodgers for reviewing this article. This work was supported by funds of the Texas Veterinary Medical Diagnostic Laboratory.
Footnotes
a.
Sigma-Aldrich Corp., St. Louis, MO.
b.
TaqGold, Promega Corp., Madison, WI.
c.
Dr. Heidi M. Hoard, Department of Pathology, Michigan State University, East Lansing, MI.
d.
GeneAmp® 9700, PerkinElmer Inc., Waltham, MA.
e.
GelRed™, Phenix Research Products, Candler, NC.
f.
VIC® and ABI Prism® 7000; Applied Biosystems Inc., Foster City, CA.