
Abstract
A PCR assay was validated for the detection of Mycoplasma hyopneumoniae in porcine lung tissue. The detection limit of the assay was 0.18 colony-forming units/g of lung sample spiked with M. hyopneumoniae. In field validation, 426 pigs from 220 cases were examined for M. hyopneumoniae infection by M. hyopneumoniae PCR and a fluorescent antibody (FA) test. In total, 103 pig lungs (24.2%) were positive in the PCR test, and 69 pig lungs (16.2%) were positive in the FA test, among which, 62 pigs were positive for both PCR and FA test. Most of the PCR-positive but FA test-negative cases had lesions compatible with M. hyopneumoniae infection. With Bayesian modeling, the diagnostic sensitivity and specificity of the PCR were determined to be 97.3% and 93.0%, respectively.
Mycoplasma hyopneumoniae causes enzootic pneumonia, which has been reported worldwide and is the most common and economically important swine disease. 14 Because of the slow growth of M. hyopneumoniae and the overgrowth of other mycoplasmas (e.g., Mycoplasma hyorhinis), isolation of this organism by culture is difficult. Other antigen or DNA detection tests include the fluorescent antibody (FA) test, immunohistochemistry (IHC), in situ hybridization, and PCR. The advantages and disadvantages of these tests have been reviewed recently. 19 The FA test has been the major antigen-identification assay; however, similar to the IHC, it may generate false-negative results if airway is not included in the tissue section. 19 In addition, false-negative results may occur if the samples are not fresh or not stored (frozen) properly. Since the early 1990s, many M. hyopneumoniae PCR assays, including single PCR or nested PCR, have been described 19 and have been found to be similar or more sensitive than the FA test. 3,18,20 Recently, 2 real-time PCR assays have been described to be highly sensitive for the detection of M. hyopneumoniae from bronchial or nasal swabs at the herd level, using both assays in combination, although the sensitivity was low when using only 1 of the assays or at the individual animal level. 7,22
In the past several years, many studies have been conducted to determine the diagnostic value of different sample types for PCR testing, and the conclusion remains indefinite. Bronchoalveolar lavage fluid samples were found to be suitable and superior to tonsil and tracheobronchial swabs, lung tissue samples, and tracheobronchial brush samples. 2,10 Nasal swabs were found to be good indicators of the presence of M. hyopneumoniae in the bronchi 17 and were more suitable than tonsil tissue samples. 17 In another study, lung tissue and nasal swabs were found not to be reliable indicators of experimentally induced M. hyopneumoniae infection, 10 and it was recommended that nasal swabs should only be used for monitoring disease status at the herd level. 13 Another report described the detection rate as higher in lung homogenates than in nasal swabs in pigs naturally infected with M. hyopneumoniae. 13 Nested PCR has been described to be a sensitive tool for the detection of M. hyopneumoniae. 6,10,13,20 However, nested PCR may be prone to cross-contamination 1 and may be unnecessary if the proper samples are used. 10 Although many M. hyopneumoniae PCRs have been validated using clinical samples, very few large-scale field validations have been reported; therefore, the information on diagnostic specificity/sensitivity is limited.
A PCR test targeting the 16S rRNA gene has been developed in nested and non-nested PCR formats and applied for detection of M. hyopneumoniae in nasal swabs. 5,6,11 This study was conducted to optimize, validate, and apply the non-nested PCR assay for the detection of M. hyopneumoniae in swine lung tissue. Since there is no perfect test (gold standard) for the detection of M. hyopneumoniae, the diagnostic specificity and sensitivity were analyzed using Bayesian rather than frequentist statistics so that the uncertainty about all parameters was modeled with probability that reflected the scientific uncertainty of the unknown quantities.
All bacteria used in this study were obtained from the American Type Culture Collection (ATCC) or were isolated and characterized at the Animal Health Laboratory (AHL), University of Guelph, Guelph, Ontario. The Mycoplasma strains were stored at −70°C in Friis broth a , 15 and other bacteria in brain heart infusion b broth containing 30% glycerol. Before use, the Mycoplasma strains were propagated in Friis broth, and other bacterial strains were cultured in trypticase soy broth. a
From the year 2000 to 2004, lung tissue samples were selected randomly from cranioventral (CV) and caudal lobes with or without pneumonic lesions from routine submissions to the AHL. The same portions of the lung samples were tested by PCR, FA test, and histopathology examination. Lung tissue DNA was extracted using a DNA extraction kit c following the manufacturer's protocol. Briefly, 3 or more pieces of tissue (total 20–25 mg) were cut from each lung and placed into a sterile safe-lock microcentrifuge (manufacturer: Eppendorf, Missisauga, Ont, Canada) 1.5-ml tube with 180 μl of lysis buffer and digested with 20 μl of proteinase K (>600 mAU/ml) in a shaking dry bath (55°C) till the tissues were completely lysed (2-4 hours or overnight). The complete homogenate was used for DNA extraction following the manufacturer's instruction exactly, except that 50 μl of elution buffer was used to elute DNA. DNA was extracted from pure cultures of Mycoplasma organisms and other bacteria using a commercial DNA extraction kit. d The PCR assay was done in a 25-μl reaction volume containing 2 mM MgCl2, e 0.2 millimoles each of deoxynucleotide triphosphate, e 0.75 U of heat-activating polymerase, f 0.2 μm of each primer, g and 2 μl of DNA extract. Primers MH649F (5′-GAG CCT TCA AGC TTC ACC AGG A-3′) and MH649R (5′-TGT GTT AGT GAC TTT TGC CAC C-3′) have been described previously. 11
Reactions were carried out in a thermocycler. h The reaction started with an initial polymerase-activating temperature of 95°C for 12 minutes, followed by 35 cycles of denaturing at 94°C for 20 seconds, annealing at 60°C for 30 seconds, elongating at 72°C for 40 seconds, and a final elongation step at 72°C for 7 minutes. After the PCR was completed, 18 μl of PCR products were electrophoresed in a 1.5% agarose gel, stained with ethidium bromide, and visualized using an ultraviolet camera. i In each PCR test run, known M. hyopneumoniae positive and negative lung tissue samples were included in the whole procedures, from sample preparation through DNA extraction and PCR amplification. In addition, M. hyopneumoniae DNA samples and pure water were also included as template and nontemplate controls.
M. hyopneumoniae on the surface of the bronchial and bronchiolar epithelium were visualized using the FA test. 21 Briefly, the lung tissue was cut into 12-mm slices and mounted on a glass slide. A drop (10 ml) of 1:80 diluted M. hyopneumoniae rabbit antiserum 15 was applied to the 1 section of the cut tissue, and 10 ml of 1:80 diluted heterologous hyperimmune rabbit serum k , 15 on the adjoining section as control. The slide was incubated at room temperature for 30 minutes then rinsed with phosphate buffered saline (PBS). After a drop of goat antirabbit fluorescein-labeled antiserum conjugate was applied to the tissue sections, the slide was incubated at room temperature for 30 minutes, then washed again in PBS before being examined under an epifluorescence microscope. The test was considered positive if micro-organisms within and/or on the surface of the airways fluoresced with M. hyopneumoniae antisera and did not fluoresce with the heterologous hyperimmune rabbit serum.
For histologic examination, sections of lung were taken from CV and caudal lobes, routinely processed and stained HE, and examined by the same pathologist. The pig was classified as having lesions compatible with M. hyopneumoniae infection if it met the following criteria: hyperplasia of bronchiolar epithelial cells, peribronchial lymphocytic cuffing, presence of moderate to large numbers of macrophages in alveoli, and perivascular lymphocytic cuffing. Mixed infection was diagnosed when more than 1 pathogen was identified by microbiology testing and/or histology lesions compatible with mixed infection were found.
Diagnostic sensitivity and specificity were calculated by Bayesian modeling, 4,9 using the freeware program Win-BUGS (http://www.mrc-bsu.cam.ac.uk/bugs). Prior distribution of model parameters was determined by obtaining expert opinion. 8,9 Pairwise and overall agreement among current diagnostic tests (FA test, histopathology, and PCR) were determined using a kappa statistic. 16
Lung tissue samples spiked with M. hyopneumoniae culture were tested to determine the analytic sensitivity and specificity of the PCR assay. A pure culture of M. hyopneumoniae strain SuiJ was propagated in Friis broth for 48 hours at 37°C. The colony-forming unit (cfu) was determined by plating 10 μl of each dilution onto Friis agar plates and counting the colonies after incubating the plates at 37°C for 48 hours. Serial dilutions of the culture were spiked in lung tissues (25 mg each) to a final concentration of 0.018 to 180,000 cfu of M. hyopneumoniae per gram of lung tissue. Among these, the samples of 0.18-180,000 cfu/g were PCR positive.
Of the 5 mycoplasma species, including Mycoplasma arginini 108, Mycoplasma flocculare MS42, Mycoplasma hyopneumoniae SuiJ, Mycoplasma hyorhinis MH31 , Mycoplasma hyosynoviae S16, and 14 other bacterial species, including Actinobacillus suis AHL110, Arcanobacterium pyogenes AHL104, Escherichia coli ATCC 25922, Erysipelothrix rhusiopathiae AHL111, Klebsiella pneumoniae AHL82, Pasteurella multocida AHL83, Pseudomonas aeruginosa ATCC27852, Proteus mirabilis AHL107, Serratia marcescens AHL86, Staphylococcus aureus ATCC25923, Staphylococcus hyicus AHL112, Streptococcus equisimilis AHL105, Streptococcus intermedius AHL106, and Streptococcus suis AHL109, only the M. hyopneumoniae strain was PCR positive; all other bacteria were PCR negative.
Total of 426 pig lung samples from 220 cases was examined for M. hyopneumoniae infection by M. hyopneumoniae PCR in parallel with an FA test. An example of the PCR results is shown in Fig. 1. In total, there were 103 pig lung samples (24.2%) positive in the PCR test and 69 pig lungs (16.2%) positive in the FA test. There were 378 samples with correlated PCR and FA test results and 48 samples with differing PCR and FA test results (Table 1). Agreement between FA test and PCR was moderate with a kappa value of 0.654. The Bayesian statistics showed that the diagnostic sensitivity and the diagnostic specificity were 97.3% and 93.0%, respectively, for the M. hyopneumoniae PCR; and the prevalence of M. hyopneumoniae was 20.2% with 95% probability interval of 16.0-24.9 (Table 2). The following prior information was used in determining the beta distributions of each parameter: M. hyopneumoniae FA test, sensitivity prior = 0.3, beta = (13.34, 29.81); specificity prior = 0.97, beta = (167.2, 42.5); for M. hyopneumoniae PCR, sensitivity prior = 0.95, beta = (329.7, 11.2); specificity prior = 0.8, beta = (19, 1); prevalence prior = 0.3, beta = (4, 10).
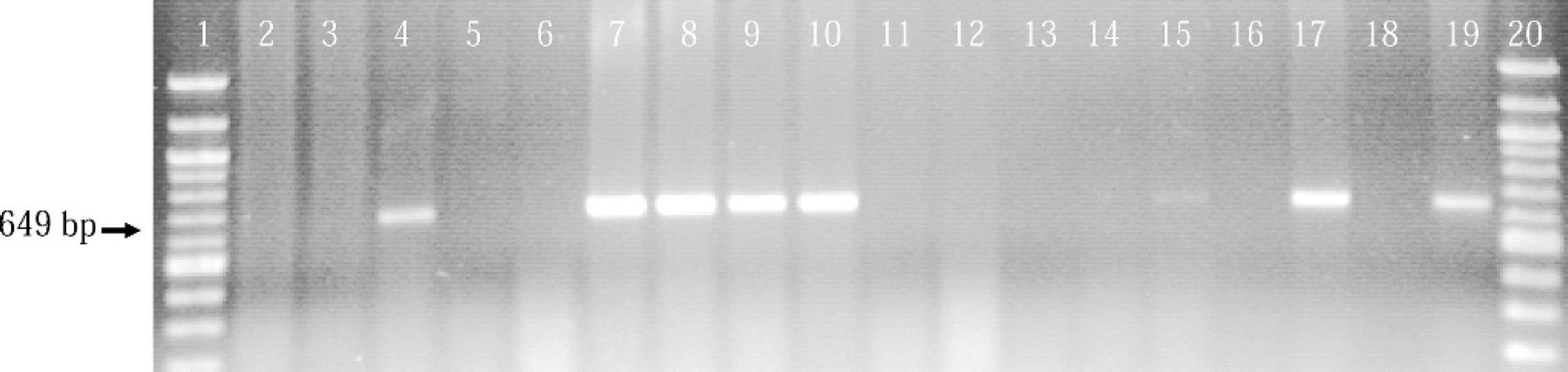
Example of M. hyopneumoniae PCR on clinical samples. Lanes 1 and 20: DNA molecular-weight marker. Lanes 2–15: clinical lung samples. Lane 16: negative control lung. Lane 17: positive control lung. Lane 18: no template, negative control. Lane 19: positive DNA control. Clinical samples in lanes 4 and 7–10 were PCR positive, and the sample in lane 15 was PCR weak positive. The positive-PCR products were 645 base pairs in size.
It has been noted that there is no true gold standard available for M. hyopneumoniae tests in a diagnostic setting. 7 Although the M. hyopneumoniae FA test is a traditional test used to diagnose infected animals, it is not a gold standard (i.e., perfect test). Therefore comparing a new, possibly more accurate test such as the M. hyopneumoniae PCR would lead to misclassification of data and test sensitivity and specificity. With Bayesian rather than frequentist statistics, the uncertainty about all parameters is modeled with probability that reflects scientific uncertainty of the unknown quantities. 9 The reason for uncorrelated results between PCR and FA tests may be that the FA test has a lower sensitivity. Among 426 samples tested, 48 samples did not have correlated PCR and FA test results, with 41 samples being PCR positive and FA test negative and 7 samples being PCR negative but FA test positive. Except for 6 samples with no histology results, most of the PCR-positive but FA test-negative samples (16/19) were found to have lesions compatible with M. hyopneumoniae infection. There were 2 samples with a positive or suspicious FA test but negative PCR. The sample with a suspicious FA test but negative PCR had no obvious mycoplasma lesion (histologic examination was not done on the case positive in the FA test but negative in PCR) (Table 3). Therefore, as displayed by the Bayesian statistical analysis, the PCR assay appeared to be more sensitive than the FA test.
PCR is an amplification method; it may detect lower numbers of M. hyopneumoniae cells compared with the antibody-antigen reaction-based FA test. In addition, the FA test requires bronchiolar epithelial cells to be intact in the samples, since M. hyopneumoniae infects the luminal surface of the bronchiolar epithelial cells. In mail-in frozen samples, if the lungs thawed during transport, the bronchiolar epithelial cells would slough and the antigen would not be detected by FA test. We tested both mail-in samples and fresh tissue from the AHL postmortem room. Among the cases with uncorrelated FA and PCR tests, 69% (18/26) were mail-in samples (Table 3). Destruction of bronchiolar epithelial cells in sick pigs may be another reason for low sensitivity of the FA test. The majority of pneumonias in pigs with uncorrelated PCR and FA test results had multiple infections with porcine respiratory and reproductive syndrome virus, swine influenza viruses, porcine circovirus type 2, M. hyopneumoniae, and other bacteria (Table 3). The co-infections may have destroyed the bronchiolar epithelial cells to the point where the number of epithelial cells were insufficient for observation in the FA test. This does not appear to have a significant impact on the PCR test. The Bayesian statistical analysis demonstrated that the PCR had lower specificity than did the FA test (Table 2). The lower specificity could be due to more false-positives than the FA test.
Comparison of PCR and fluorescent antibody (FA) tests for the detection of M. hyopneumoniae in lung.

Choice of prior information is also important in Bayesian analysis, and the input for PCR specificity was lower than that for the FA test. Additionally, diagnostic sensitivity and specificity are often inversely related.
Diagnostic sensitivity and specificity for M. hyopneumoniae fluorescent antibody (FA) test and PCR, using Bayesian statistics (n = 426).

PI = probability interval.
Cases with uncorrelated Mycoplasma hyopneumoniae PCR and fluorescent antibody (FA) test results.
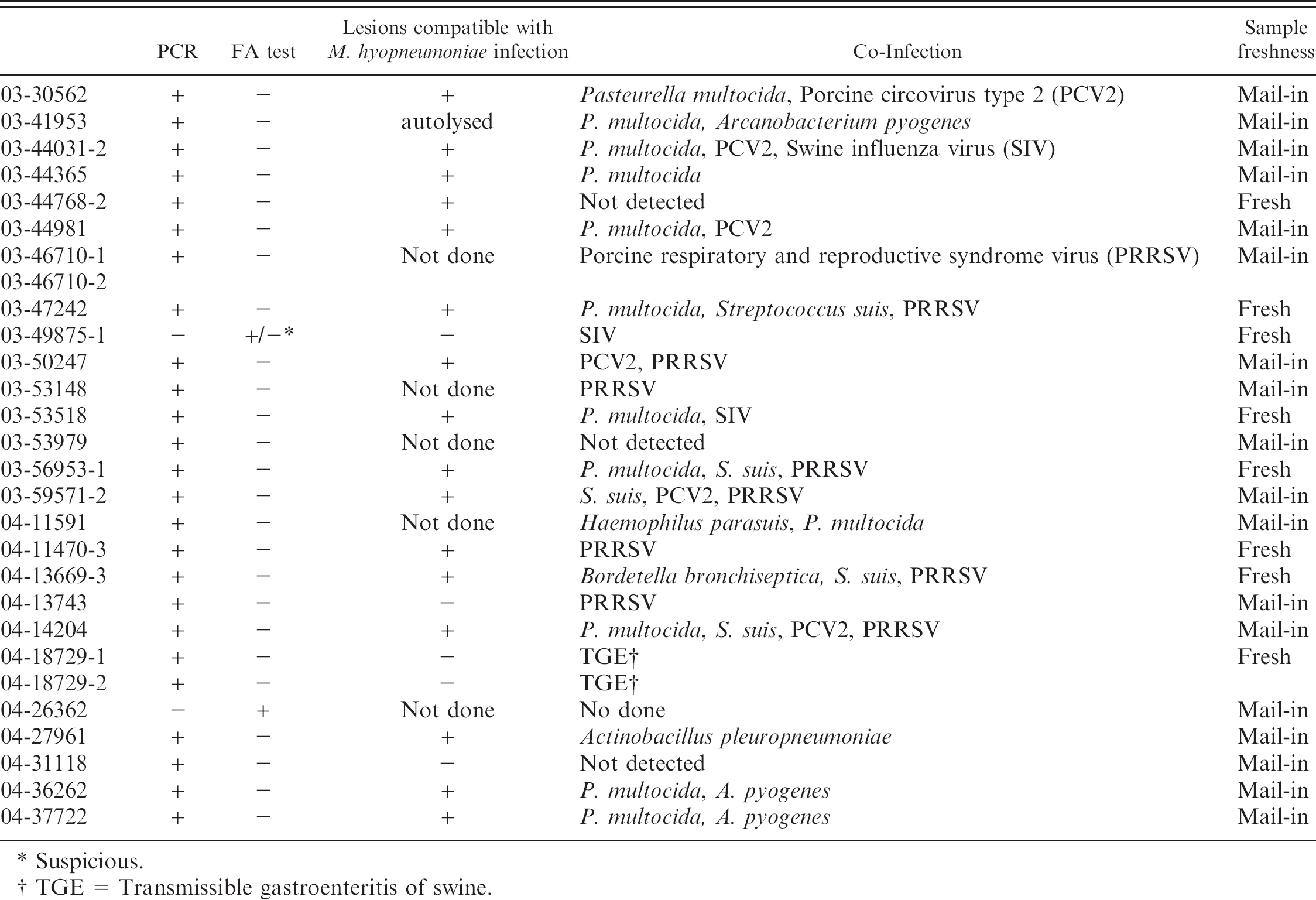
Suspicious.
TGE = Transmissible gastroenteritis of swine.
Biologic factors such as disease attributes and sampling techniques will also affect the sensitivity and specificity of tests. The positive predictive value (PPV) is the probability that given a positive test, the animal actually has the disease. It is dependent upon prevalence; it actually increases if the test has a high specificity (no false positives), which accounts for the higher PPV of the FA test compared with the PCR. If cost is not an issue, we would recommend the use of PCR and FA tests in combination to increase their sensitivity or specificity.
The PCR assay was originally reported for testing swab samples. The detection limit of the PCR was described to be 5 cfu, determined by testing the lysed cells directly without other DNA extraction treatment. 11 This study adapted and optimized the PCR for testing of lung tissue samples. The PCR detection limit was improved in this study by using a commercial DNA extraction kit, which has been shown to be superior to some other DNA extraction methods. 12 In addition, the use of hot-start PCR in this study reduced the competition from nonspecific amplification, which may be another reason for increased PCR sensitivity. We found that carry-over contamination could be prevented by using the proteinase K digestion method to replace the mechanical homogenization (data not shown). Although the problem of mechanical homogenizer causing cross-contamination has not been described previously, we found from our experience that it was very difficult to clean or degrade the DNA in the mechanical tissue homogenizer, which can cause PCR cross-contamination. In addition, we compared nested and non-nested format M. hyopneumoniae PCRs in our preliminary experiments, and we experienced cross-contamination frequently in the nested PCR. Therefore, we decided not to use the nested PCR.
In conclusion, the PCR optimized and validated in this study is sensitive and specific and is a better alternative to the FA test for the detection of M. hyopneumoniae in lung tissue samples.
Acknowledgements. This project was financially supported in part by Ontario Pork and the Ontario Ministry of Agriculture, Food and Rural Affairs.
Footnotes
a.
Difco, Detroit, MI.
b.
Becton Dickinson, Cockeysville, MD.
c.
Qiagen DNeasy Tissue kit, Qiagen, Mississauga, Ontario, Canada.
d.
Instagen Matrix, BioRad, Mississauga, Ontario, Canada.
e.
PE Applied Biosystems, Mississauga, Ontario, Canada.
f.
AmpliTaq Cold polymerase, PE Applied Biosystems, Mississauga, Ontario, Canada.
g.
Molecular Supercenter, University of Guelph, Guelph, Ontario, Canada.
h.
PCR System 9600, PE Applied Biosystems, Mississauga, Ontario, Canada.
i.
Gel Documentation System, BioRad, Mississauga, Ontario, Canada.
j.
Rabbit antiserum against M. hyopneumoniae strain SuiJ (Aarhus University, Denmark), Animal Health Laboratory, University of Guelph, Guelph, Canada.
k.
Rabbit antiserum against Mycoplasma hyorhinis strain 10118 (National Collection of Type Cultures, London, England), Animal Health Laboratory, University of Guelph, Guelph, Canada.