
Abstract
Purpose
The abnormal function and survival of chondrocytes result in articular cartilage failure, which may accelerate the onset and development of osteoarthritis (OA). This study is aimed to investigate the role of LINC01094 in chondrocyte apoptosis.
Methods
The viability and apoptosis of lipopolysaccharide (LPS)-induced chondrocytes were evaluated through CCK-8 assay and flow cytometry analysis, respectively. The expression levels of LINC01094, miR-577 and MTF1 were detected by qRT-PCR. Dual luciferase reporter tests were implemented for the verification of targeted relationships among them. Western blotting was employed to measure the levels of pro-apoptotic proteins (Caspase3 and Caspase9).
Results
The viability of LPS-induced chondrocytes was overtly promoted by loss of LINC01094 or miR-577 upregulation, but could be repressed via MTF1 overexpression. The opposite results were observed in apoptosis rate and the levels of Caspase3 and Caspase9. LINC01094 directly bound to miR-577, while MTF1 was verified to be modulated by miR-577. Both LINC01094 and MTF1 were at high levels, whereas miR-577 was at low level in OA synovial fluid and LPS-induced chondrocytes. Furthermore, the highly expressed miR-577 abolished the influences of MTF1 overexpression on LPS-induced chondrocytes.
Conclusions
Silencing of LINC01094 represses the apoptosis of chondrocytes through upregulating miR-577 expression and downregulating MTF1 levels, providing a preliminary insight for the treatment of OA in the future.
Keywords
Introduction
Osteoarthritis (OA), as the most common form of arthritis, can generally affect lower extremity joints such as the hip and knee. 1 It is a leading cause of disability among middle-age and older adults worldwide, leading to function loss, pain and the decreased quality of life. 2 The most prominent features of OA are joint inflammation and the progressive degeneration of articular cartilages. 3 Chondrocyte, as the sole cell type among articular cartilages, is considered to be involved in the degenerative processes. Although some advancements have been made in alleviating inflammation and pain in patients with OA, the development of OA is still not modified. Therefore, an in-depth investigation in how to suppress chondrocyte apoptosis is indispensable to develop a potentially adjuvant therapy for OA in clinical practice.
Increasing investigations have shown that the onset and development of OA involve the modulation of various transduction pathways and genes, including long non-coding RNA (lncRNA), a series of RNA longer than 200 nucleotides that do not encode proteins.4,5 For instance, Wang et al. uncovered a pronounced upregulation of lncRNA CIR in OA patients, while silencing of lncRNA CIR dramatically attenuates the degeneration of articular cartilage via regulating autophagy-related proteins. 6 Sun et al. build an OA cell model via interleukin (IL)-1β treatment in human chondrocytes SW1353 and demonstrated that lncRNA XIST is overexpressed in IL-1β-challenged SW1353 cells. 7 In another case, Hu et al. found that downregulation of lncRNA H19 can ameliorate the damage of lipopolysaccharide (LPS) to C28/I2 cells, namely the inhibiting effect on apoptosis and the promoting impact on cell growth. 8 Notably, LINC01094, a lncRNA located at chromosome 4, has been verified to play oncogenic role in the tumorigenicity of human cancers.9,10 Nonetheless, accurate knowledge on the function of LINC01094 in the occurrence and development of OA is relatively rare.
MicroRNAs (miRNAs) are a category of ncRNAs that modulate the expression levels of target genes to exert regulate functions. Numerously previous data have shown strong associations between hundreds of miRNAs and genes relevant to OA pathogenesis. Specifically, the dysregulation of miRNAs is involved in the cartilage degeneration-related pathologic phenomena such as apoptosis, inflammation and autophagy. 11 For examples, decreased expression of miR-26a and miR-26b in cartilage contributes to the development of OA. 12 Both miR-140-5p and miR-140-3p are confirmed to be downregulated in OA patients and this event aggravates the severity of OA. 13 miR-214-3p, also a reduced miRNA in OA cartilage, significantly induce chondrocyte apoptosis and thus promote OA progression. 14 As for miR-577, it is reported to be a regulator of cell proliferation and apoptosis in several human cancers.15,16 Additionally, as the public data indicated, lncRNAs mediating miRNA sponges are of great importance in OA progression, such as lncRNA MALAT1-miR-150-5p, 17 lncRNA MFI2-AS1-miR-130a-3p 18 and lncRNA MEG3-miR-16. 19 More importantly, miR-577 is verified to be mediated by lncRNA DANCR to be participated in OA pathology. 20 However, whether lncRNA LINC01094-miR-577 network has an effect on OA pathogenesis is still poorly understood.
The current investigation preliminarily ascertained the role of lncRNA LINC01094 in OA progression. At the same time, this study for the first time pointed out that LINC01094 knocking-down inhibits the onset and development of OA through mediating miR-577/metal-regulatory transcription factor 1 (MTF1) axis.
Methods
Cells and reagents
From Procell (Wuhan, China), human normal chondrocytes were purchased. Fetal bovine serum (FBS) was from Gibco (Grand Island, NY, USA). DMEM/F12 medium was obtained from Hyclone Technologies (Logan, UT, USA). Penicillin-Streptomycin Solution was available from Beyotime Biotechnology (Shanghai, China). LPS was bought from Sigma Aldrich (St Louis, MO, USA). LINC01094-siRNA-1/-2/-3 and the negative control (NC siRNA), overexpression of MTF1 (pcDNA-MTF1) and the empty vector (pcDNA-NC), as well as miR-577 mimics/NC mimic were synthesized by VectorBuilder (Guangzhou, China). Dojindo Laboratories (Shanghai, China) provided the CCK-8 Kit. Annexin V-FITC/PI Apoptosis Detection Kit was obtained from Vazyme (Nanjing, China). For western blotting, primary antibodies Caspase3, Caspase9 and GAPDH were got from Proteintech (Wuhan, China). Abcam (Cambridge, UK) provided the HRP-conjugated secondary antibody. Lipofectamine 3000 and Trizol reagent were procured from Invitrogen (Carlsbad, CA, USA). Hifair® Ⅱ 1st Strand cDNA Synthesis SuperMix and Hieff® qPCR SYBR Green Master Mix were obtained from Yeasen Biotechnology (Shanghai, China).
OA synovial fluid collection
A total of 10 OA patients were enrolled in our hospital, and 10 healthy people were served as the controls. Synovial fluid from knee joints of OA cases and healthy controls was obtained. The collected synovial fluid samples were immediately stored in liquid nitrogen until used. All the individuals had provided the informed consent. This study obtained the approval of our hospital’s ethics committee (approval No.KY-2022-335) and conformed to the guidelines of the Declaration of Helsinki.
Cell culture and treatment
Human normal chondrocytes were cultured in DMEM/F12 with an added 10% FBS and 1% Penicillin-Streptomycin Solution, keeping in the environmental conditions of 5% CO2 at 37°C. As previously described, 8 5 µg/mL of LPS was used to treat normal chondrocytes for 12 h to establish OA cell model.
Cell transfection
Based on Lipofectamine 3000 reagent’s instructions, the transfection experiments were performed. In brief, LINC01094-siRNA-1/-2/-3, NC siRNA, miR-577 mimics, NC mimic, pcDNA-MTF1 and pcDNA-NC were transfected individually into chondrocytes as needed, and incubated for 48 h. The cells were then collected for the subsequent experiments.
RNA isolation and qRT-PCR
Real-time PCR Primer synthesis list.

Western blotting
The protein levels of Caspase3 and Caspase9 in LPS-induced chondrocytes were determined through western blotting. Briefly, protein was extracted from LPS-induced chondrocytes using RIPA lysis buffer. Subsequently, 100 μg of protein samples was electrophoresed and separated via a 10% SDS-PAGE, followed by transfer onto the PVDF membranes. The membranes were blocked with 5% nonfat milk at room temperature, and then incubated with primary antibodies anti-Caspase3 (dilution in 1:500), anti-Caspase9 (dilution in 1:600), and anti-GAPDH (dilution in 1:5000). After overnight incubation at 4°C, an HRP-conjugated secondary antibody (dilution in 1:5000) was used and at the same time, the membrane was incubated at 25°C for 2 h. A BioSpectrum 600 Imaging System (UVP, CA, USA) was used for the detection of protein signals.
CCK-8 assay
CCK-8 assay was employed to measure the viability of chondrocytes. Chondrocytes (4 × 103 cells/well) were cultured in 96-well plates. After 12, 24, 36 and 48 h of incubation, 10 μL of CCK-8 solution was added to the respective wells. The duration of the reaction was 3 h. The viability of chondrocytes was calculated using a BIOTEK microplate reader (Vermont, USA) at 450 nm.
Flow cytometry analysis
The apoptosis of chondrocytes was evaluated using an Annexin V-FITC/PI Apoptosis Detection Kit, in accordance with the manufacturer’s protocol. In brief, chondrocytes (2 × 105) were re-suspended in binding buffer (500 μL), followed by staining with PI and Annexin V-FITC (both 5 μL) at 4°C for 15 min in the dark. Subsequently, cell apoptosis was assessed using a FACScan flow cytometer (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
Dual luciferase reporter assays
The MTF1 and LINC01094 sequences containing miR-577 binding sites were combined to pGL3 vectors to construct MTF1-wild type (wt), LINC01094-wt, MTF1-mutant (mut), and LINC01094-mut. Thereupon, miR-577 mimics and NC mimic together with the above constructed vectors were transfected into chondrocytes using Lipofectamine 3000 reagent. After incubation of 24 h, the relative luciferase activities were determined via a Luciferase reporter assay system (Promega, Madison, WI, USA).
Statistical analysis
Data were indicated in the form of mean ± standard deviation and statistically analyzed using SPSS 20.0 software. Both t-test and ANOVA plus Tukey’s post-hoc test were done for the comparison among groups. p < .05 was counted as statistically significant.
Results
LINC01094 knocking-down inhibits the apoptosis of chondrocytes
The expression levels of LINC01094 in OA patients were preliminarily detected. As manifested in Figure 1(a), it was overtly increased in OA synovial fluid relative to normal synovial fluid (p < .001). Then, we found that LINC01094 expression in LPS-induced chondrocytes was also significantly upregulated compared to that in normal chondrocytes (Figure 1(b), P < .001). Either LINC01094 siRNA-1/-2/-3 or NC siRNA was transfected into chondrocytes to verify the transfection efficiency. As illustrated in Figure 2(a), LINC01094 expression levels were significantly decreased following transfection (p < .01). We selected LINC01094 siRNA-2 for the succeeding experiments due to its relatively high transfection efficiency (p < .001). We demonstrated that the amounts of LINC01094 were abnormally reduced by transfection of LINC01094 siRNA in the presence of LPS (Figure 2(b), P < .01). Meanwhile, CCK-8 assay and flow cytometry analysis indicated that the silenced LINC01094 could dramatically induce cell viability (Figure 2(c), P < .05) but suppress the apoptosis of chondrocytes (Figure 2(d), P < .001). Additionally, we further found that knockdown of LINC01094 repressed the levels of pro-apoptotic proteins (Caspase3 and Caspase9) (Figure 2(e), P < .001). Pronounced upregulation of LINC01094 is observed in OA tissues and LPS-induced chondrocytes. The expression of LINC01094 in (a) OA synovial fluid or (b) LPS-induced chondrocytes was detected via qRT-PCR. ***p < .001. Knocking-down LINC01094 prohibits the apoptosis of chondrocytes. LINC01094 expression in (a) chondrocytes or (b) LPS-induced chondrocytes. The viability (c) and apoptosis (d) of LPS-induced chondrocytes. (e) The protein levels of Caspase3 and Caspase9 after transfection. *p < .05, **p < .01, ***p < .001.

LINC01094 binds to miR-577
LncRNAs generally serve as miRNA sponges to exert regulatory functions in OA progression .17,18 Through StarBase software, the underlying binding site of miR-577 with LINC01094 was depicted (Figure 3(a)). Dual luciferase reporter assays confirmed that miR-577 mimics could overtly decrease the luciferase activity of LINC01094 wt reporter vector in chondrocytes (Figure 3(b), P < .001), but the luciferase activities between the LINC01094 mut + NC mimic and LINC01094 mut + miR-577 mimics groups had no significant changes. The results of qRT-PCR uncovered that miR-577 expression was lowly expressed in OA patients than that in normal patients (Figure 3(c), P < .001). Relative low levels of miR-577 were also determined in chondrocytes exposed to LPS (Figure 3(d), P < .001). Additionally, we demonstrated that miR-577 expression in LPS-induced chondrocytes could be remarkably elevated after LINC01094 siRNA transfection (Figure 3(e), P < .001). LINC01094 binds to miR-577. (a) Predicted binding site between LINC01094 and miR-577. (b) The luciferase activities in chondrocytes. miR-577 expression in (c) OA synovial fluid or (d) LPS-induced chondrocytes. (e) miR-577 expression in LPS-induced chondrocytes after transfection. *p < .05, ***p < .001.
The apoptosis of chondrocytes is repressed by miR-577 upregulation
miR-577 mimics or NC mimic was transfected into LPS-induced chondrocytes to explore the influences of miR-577 during the progression of OA in vitro. As shown in Figure 4(a), the expression levels of miR-577 were undoubtedly increased after miR-577 mimics transfection (p < .001). Upregulation of miR-577 exerted obviously promoting impacts over cell viability, with a time-dependent manner (Figure 4(b), P < .01). As expected, the apoptosis rate of chondrocytes in the presence of LPS could be markedly repressed under the conditions of miR-577 overexpression (Figure 4(c), P < .001). Meanwhile, upregulation of miR-577 also remarkably suppressed the protein levels of Caspase3 and Caspase9 in LPS-induced chondrocytes (Figure 4(d), P < .001). The apoptosis of chondrocytes is repressed by miR-577 upregulation. (a) miR-577 expression in LPS-induced chondrocytes. The viability (b) and apoptosis (c) of LPS-induced chondrocytes. (d) The protein levels of Caspase3 and Caspase9 in LPS-induced chondrocytes. **p < .01, ***p < .001.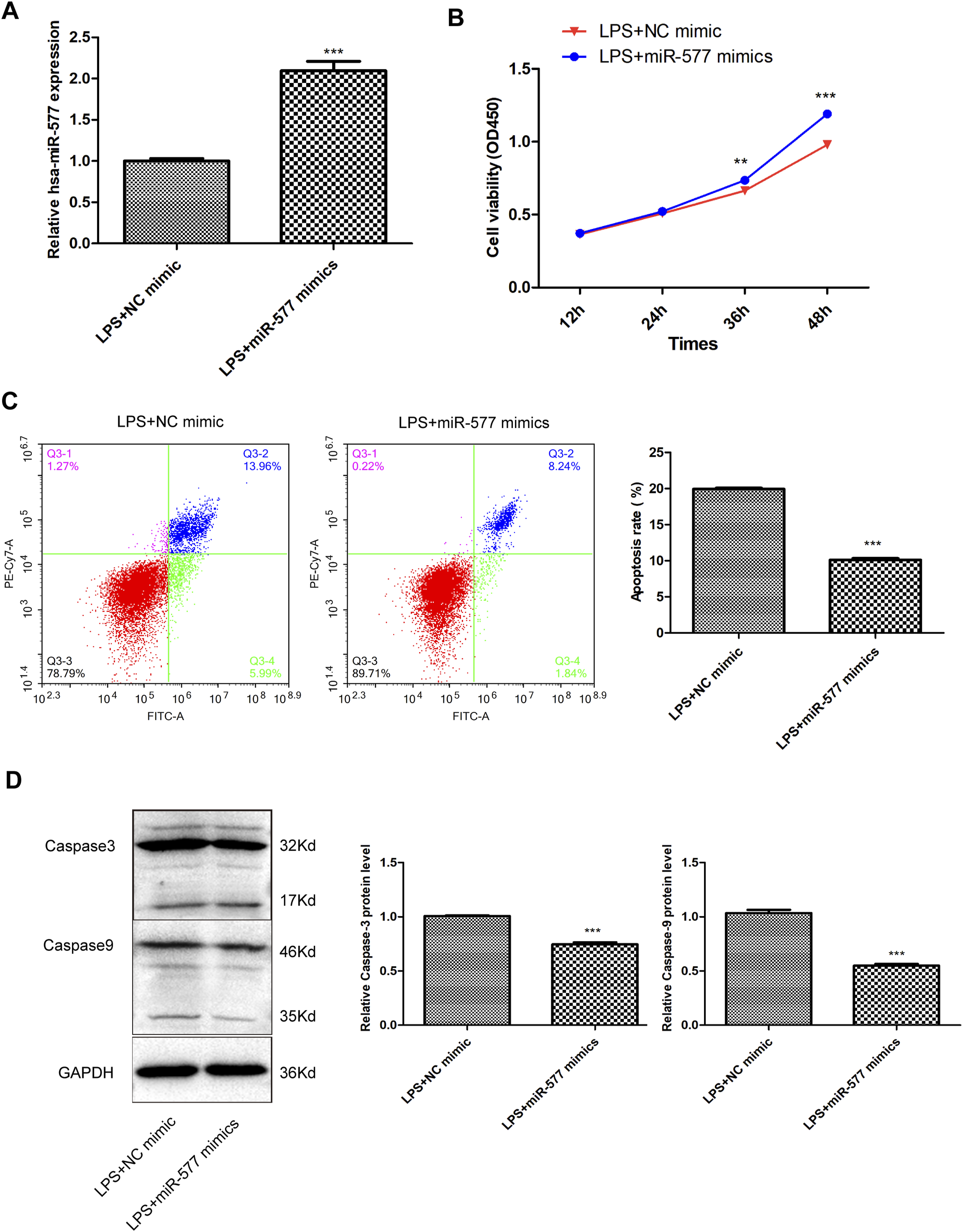
MTF1 is a direct target of miR-577
Based on TargetScan software, an underlying miR-577 and MTF1 binding site was shown (Figure 5(a)). In chondrocytes, luciferase reporter assays were employed to confirm this. As indicated in Figure 5(b), it was shown that combination of MTF1 wt with miR-577 mimics markedly reduced the luciferase activities (p < .001). But for those between MTF1 mut/NC mimic and MTF1 mut/miR-577 mimics groups, there were no obvious changes. Besides, high amounts of MTF1 were simultaneously determined in OA patients and LPS-induced chondrocytes, compared to the corresponding controls (Figure 5(c) and (d), P < .001). Additionally, we found that the expression levels of MTF1 in LPS-induced chondrocytes were overtly downregulated following transfection of LINC01094 siRNA or miR-577 mimics (Figure 5(e) and (f), P < .05). MTF1 is considered as a direct target of miR-577. (a) Predicted binding site between miR-577 and MTF1. (b) The luciferase activities in chondrocytes. MTF1 expression in (c) OA synovial fluid or (d) LPS-induced chondrocytes. (e)-(f) MTF1 expression in LPS-induced chondrocytes after transfection. *p < .05, ***p < .001.
Upregulation expression of miR-577 abolishes the influences of MTF1 overexpression on LPS-induced chondrocytes.
Following transfection of pcDNA-MTF1, the expression levels of MTF1 in LPS-induced chondrocytes were significantly elevated (Figure 6(a), P < .001), while transfection of miR-577 mimics to some extent reduced the enhancement effects of pcDNA-MTF1 introduction on MTF1 expression (p < .01). For cell viability, it was overtly repressed by the ectopic expression of MTF1 and gradually restored following miR-577 mimics transfection (Figure 6(b), P < .01). We further found that MTF1 overexpression distinctly accelerated the apoptosis rate of LPS-induced chondrocytes (Figure 6(C), P < .001), while after transfection of miR-577 mimics, the apoptosis rate was slowed down to some extent (p < .01). Meanwhile, similar results were observed in the levels of pro-apoptotic proteins (Caspase3 and Caspase9) (Figure 6(d), P < .001). Upregulation expression of miR-577 abolishes the influences of MTF1 overexpression on LPS-induced chondrocytes. (a) MTF1 expression in LPS-induced chondrocytes after transfection. The viability (b), apoptosis rate (c) and pro-apoptotic protein levels (d) of LPS-induced chondrocytes following transfection. **p < .01, ***p < .001. ##p < .01, ###p < .001.
Discussion
OA is a prevalent arthritis emerged from chondrocyte apoptosis. It is estimated that there are approximately 250 million people suffered from OA worldwide and the government costs more than 300 billion dollars annually for the treatment of OA patients. OA has become a heavy burden on public health all over the world. In the current research, a novel molecular mechanism that inhibits OA progression was uncovered, indicating that silencing of LINC01094 suppresses the apoptosis of chondrocytes via miR-577/MTF1 axis.
Recently, growing reports have displayed that the dysregulation of lncRNAs plays a prominent part across OA progression. For instance, lncRNA HOTAIR expression is at high level in OA cartilage, and its overexpression has overtly promoting impacts on chondrocyte apoptosis and degradation of extracellular matrix (ECM). 21 Pronounced upregulation levels of lncRNA FOXD2-AS1 are observed in OA chondrocytes, which is considered to be significantly associated with ECM degradation and inflammatory responses. 22 In another report, Li et al. indicated that OA chondrocytes with high expression of lncRNA PVT.1 have relatively high apoptosis rate. 23 Similar to the previous works, we demonstrated that LINC01094 expression was also at high levels in OA patients and LPS-induced chondrocytes. Moreover, our loss-of-function experiments further pointed out its repressive effects on the apoptosis of LPS-induced chondrocytes when it was silenced. These results evidenced that LINC01094 acts as a pathogenic candidate to mediate OA development.
As published studies indicated, pronounced decrease of miR-577 expression levels is observed in a series of human cancers. Low amounts of miR-577 confer neoplastic cells resistance to apoptosis, thus aggravating cancer development. However, contrary to the pathological role of apoptosis in cancer cells, chondrocyte reduction or apoptosis can result in the failure of appropriately maintaining the structure of articular cartilage, eventually accelerating the progression of OA. 24 In this report, our research data also uncovered a pronounced low expression of miR-577 in OA synovial fluid and LPS-treated chondrocytes, relative to the respective controls. We speculated that downregulation of miR-577 may be correlated with the development of OA via promoting chondrocyte apoptosis. To validate this hypothesis, miR-577 mimics or NC mimic was transfected into LPS-treated chondrocytes. Through CCK-8 assay, flow cytometry analysis, and western blotting experiments, we demonstrated that overexpression of miR-577 could promote cell viability and suppress apoptosis in LPS-induced chondrocytes. These results implied the importance of miR-577 in OA pathobiology. Additionally, our study further identified that LINC01094 could bind to miR-577 in OA and at the same time, miR-577 expression levels were negatively modulated by LINC01094. Evidence has pointed out that many lncRNAs can act as miR-577 sponges across the progression of OA and cancer.20,25 As we have confirmed the roles of LINC01094 and miR-577 in the apoptosis of LPS-induced chondrocytes, we therefore believed that LINC01094 knocking-down suppresses chondrocyte apoptosis via upregulating miR-577 expression.
Metallothionein (MT), encoded by a family of genes located in chr 16q13, is a kind of crucial protein strongly associated with the intracellular handling of metal ions. 26 MTF1, a highly conserved zinc finger protein, generally serves as a positive mediator to induce the expression of MT encoding genes. 26 It is usually at high levels across human cancers, and is recognized as a crucial oncogene in human cancers. Not only that, MTF1 has been shown to be an important transcription factor to mediate the expression of zinc/ZIP8-induced catabolic factors, thus altering the pathogenesis of OA. 27 Herein, high levels of MTF1 were identified both in OA patients and LPS-induced chondrocytes. These results supported that the abnormal expression of MTF1 is indeed associated the development of OA. Meanwhile, the results that the ectopic expression of MTF1 could repress cell viability and induce apoptosis in LPS-induced chondrocytes further validated this conclusion. Previous works have manifested that lncRNA-miRNA-mRNA-based network is of great importance in OA pathogenesis.28,29 Interestingly, MTF1 has been proposed to be a target gene of miR-577 in OA in this study. Based on this prediction, we speculated that MTF1 may act as a pathogenic factor to be involved in LINC01094-miR-577-mediated OA development. As expected, MTF1 expression in LPS-induced chondrocytes was markedly elevated by LINC01094 silencing or miR-577 upregulation. Moreover, upregulation of miR-577 could attenuate the repressive effects on cell viability or the promoting impacts on apoptosis caused by MTF1 overexpression in LPS-induced chondrocytes. Collectively, these findings to some extent uncovered that silencing of LINC01094 may ameliorate the progression of OA via modulating miR-577/MTF1 axis.
The limitations of this study should not be disregarded. Firstly, the total sample size of OA patients in this study may be relatively small, and the number of enrolled patients with OA still needs to be enlarged in the future. Secondly, the interaction between LINC01094 and miR-577 in LPS-induced chondrocytes should be elucidated. Thirdly, an OA animal model should be established to explore the relationships among LINC01094, miR-577, and MTF1.
Conclusion
To recapitulate briefly, we for the first time indicated the role of LINC01094 in the pathogenesis of OA. At the same time, its detailed action mechanism was also uncovered, namely LINC01094 knocking-down synergistic interaction with miR-577/MTF1 pathway to protect against OA development. These findings have certain groundbreaking significance for the clinical treatment of OA.
Footnotes
Authors’ contributions
YZX conceived and designed the study. FRH and ZLS conducted most of the experiments and data analysis. FRH wrote the manuscript. JY and XZZ participated in collecting data and helped to draft the manuscript. All authors are in agreement with the content of the manuscript and take responsibility for the contents of the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical statement
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.