
Abstract
The way cancer cells adapt to microenvironment is crucial for the success of carcinogenesis, and metabolic fitness is essential for a cancer cell to survive and proliferate in a certain organ/tissue. The metabolic remodeling in a tumor niche is endured not only by cancer cells but also by non-cancerous cells that share the same microenvironment. For this reason, tumor cells and stromal cells constitute a complex network of signal and organic compound transfer that supports cellular viability and proliferation.
The intensive dual-address cooperation of all components of a tumor sustains disease progression and metastasis. Herein, we will detail the role of cancer-associated fibroblasts, cancer-associated adipocytes, and inflammatory cells, mainly monocytes/macrophages (tumor-associated macrophages), in the remodeling and metabolic adaptation of tumors.
Keywords
Cancer cells metabolic fitness is crucial for cancer establishment
Metabolism is undoubtedly one of the fields of knowledge that is currently booming. In our opinion, it will certainly be a niche of scientific discoveries that will allow the evolution of the knowledge and treatment of diseases, namely cancer. The metabolic adaptation of cancer cells not only permits the development and establishment of a tumor in a certain microenvironment but also conditions the response to the therapy.
Metabolic adaptation cannot be assessed exclusively from the cancer cell point of view but should also consider the contribution of normal (non-cancerous) cells in the same tissue or organ. Metabolism does not consist solely on the intracellular network that shares and provides organic compounds among the various chemical reactions that make up the metabolic dynamics. It also encompasses the extracellular organic and signaling molecules that supplement and mediate stimuli, which regulate the entire metabolic functioning of a cell. In cancer, the surrounding cells (fibroblasts, endothelial cells, immune cells, and adipocytes) influence the tumor microenvironment, the tumor cell biology and metabolism. In fact, in the metabolic interactions between tumor and stroma, the tumor cells take advantage of the metabolic substrates (e.g. glutamine, lactate, and fatty acids) from local and/or stromal sources.1–4 Thus, the tumor microenvironment is composed and depends on the functioning of several types of cells (malignant and normal) that continuously share organic and signaling molecules, cooperating and making possible the entire system’s survival. We will include in this review both solid and hematological tumors, as the former despite the spread in the blood stream are also generated and maintained in an organic context, such as bone marrow, spleen, and lymph nodes.
The connective tissue, which is the most part of the stroma, plays an important role in the sustainability of a tumor and cancer cells homing, being cancer-associated fibroblasts (CAFs), cancer-associated adipocytes (CAAs) and inflammatory cells, mainly monocytes/macrophages (tumor-associated macrophages (TAMs)) important players in cancer development.
Fibroblasts
Fibroblasts are the major component of the tumor stroma, playing an important role in cancer progression through their molecular cooperation with cancer cells. CAFs and tumor cells symbiosis allows a supportive and enriched microenvironment, important for tumor survival, proliferation, migration, and chemoresistance5,6 (Figure 1). Despite the molecular mechanisms being not fully known, this cooperation has been described in different cancer contexts, such as pancreatic, colon, ovarian, leukemia, and breast cancer.7–13 Also, CAFs are able to support angiogenesis and metastasis by promoting matrix remodeling and epithelial-to-mesenchymal transition (EMT).14–16

CAFs are reprogrammed by cancer cells and tumor microenvironment. Cancer cells produce cytokines (interleukine-6 (IL-6)), chemokines (chemokine (C–X–C motif) ligand 2 (CXCL2)), and growth factors (tumor growth factor (TGF) and platelet-derived growth factor (PDGF)) to stimulate the transformation of normal fibroblasts (NFs) into activated cancer-associated fibroblasts (CAFs).24–26 CAFs reprogramming involves the loss of caveolin 1 (Cav-1) and an increase in the levels of α-smooth muscle actin (α-SMA) and tenascin. 23 CAFs will control tumor growth, metastasis, and angiogenesis through the secretion of TGFβ, IL-6, CXCL12, and chitinase-3-like-1 (CHI3L1).27–29
Fibrotic tissue has a role in cancer promotion and progression
In a normal and physiologically regenerative process, the wound site is strongly enriched in activated normal fibroblasts (NFs), a reversible process that confers an enhancement of contractile, synthetic, and metabolic properties.17–20 Activated NFs have an important and recognized role in acute injury due to their ability to produce collagen, fibronectin, cytokines, and growth factors. 19
NFs and CAFs are distinct cell types with different molecular markers and functional plasticities; hence, the close contact of CAFs and cancer cells within a tumor is essential for a favorable pro-carcinogenic microenvironment.17–20
In comparison to NFs, CAFs display higher levels of pro-tumorigenic features, and they have lower contractility and an increased survival, proliferation, and extracellular matrix (ECM) remodeling capacity. Although CAFs retain the capacity of synthesizing and secreting collagen and fibrous proteins (e.g. reticulin, elastin, and glycoproteins).17,21–23 In vitro studies in colon 9 and pancreatic cancer7,11 described that cancer cell lines increase their proliferation rate upon exposure to CAFs-conditioned media. CAFs also secrete chemokines to promote angiogenesis in a paracrine-dependent manner. Chemokine (C–X–C motif) ligand 2 (CXCL2), also known as stromal cell–derived factor-1 (SDF-1), secreted by CAFs binds to C–X–C chemokine receptor type 4 (CXCR-4) in tumor cells, increasing CXCL12/CXCR4 signaling, which promotes proliferation and angiogenesis. 27 Recently, chitinase-3-like protein 1 (CHI3L1) secreted by CAFs has been implicated in the promotion of tumor progression and metastasis of breast carcinomas 28 through an unknown mechanism (Figure 1).
Besides their differences, NFs and CAFs still share their activation process which requires the intervention of several elements, such as transforming growth factor beta 1 (TGF-β1), platelet-derived growth factor (PDGF), cyclooxygenase-2 (COX-2), CXCL12, and interleukin-6 (IL-6)24–26,30–32 (Figure 1).
CAFs reprogramming involves the loss of caveolin 1 (Cav1) and an increase in the levels of α-smooth muscle actin (α-SMA) and tenascin 23 (Figure 1). Recently, Busch et al. 33 reported a new member of aldehyde dehydrogenase (ALDH) family, implicated in CAF transformation and consequent secretion of angiogenic and chemotactic growth factors. In addition, CAFs are enriched in growth differentiation factor 5 (GDF5), a member of TGF-β1/bone morphogenetic protein (BMP) family, that supports the migration, invasion, and progression of pancreatic cancer cells. 29
CAFs play a role in cancer metabolic fitness
The higher proliferation observed in CAFs is sustained by an increase in glycolysis and autophagy in order to mobilize internal sources of nutrients, providing to the Krebs cycle (tricarboxylic acid cycle (TCA)) with metabolic intermediates. The glycolytic switch observed in CAFs that are needed to support the metabolic adaptation of cancer cells, even in normoxia conditions, may be driven by a decrease in oxygen availability, hypoxia-inducible factor-1 alpha (HIF-1α) stabilization, transforming growth factor β (TGF-β) and PDGF signaling, or reactive oxygen species (ROS)-mediated loss of caveolin-130–32,34–38 (Figure 1).
IL-6 has been described as a putative intervenient in a metabolic switch of CAFs for glycolysis by increasing the expression of glycolytic enzymes such as phosphofructokinase, hexokinase, and fructose-2,6-biophosphatase via signal transducer and activator of transcription 3 (STAT3) activation. 39 Also, TGF-β1-/PDGF-mediated CAF differentiation induces a decrease in the expression of Krebs cycle enzymes concomitant with an increase in glucose uptake and glycolysis.40,41 In metabolically reprogrammed CAFs, the downregulation of isocitrate dehydrogenase 3a (IDH3a) leads to a decrease in α-ketoglutarate (α-KG) and consequently decrease in the stabilization of HIF-1α protein, which in turn promotes glycolysis and inhibits oxidative phosphorylation by upregulating NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 4-like 2 (NDUFA4L2), even under normoxic conditions. 41 Despite the precise mechanism driving the increase in glycolysis in CAFs is not fully understood, it has been demonstrated that CAFs from breast cancer patients with poor prognosis express higher levels of glycolytic enzymes. 10
Cancer cells take advantage of the resulting metabolites from the altered CAF metabolism (Figure 2). A good example of the constant and dynamic metabolic crosstalk between CAFs and tumor cells is lactate, the final metabolite of glycolysis, which is produced and exported by CAFs and further imported and consumed by cancer cells. CAF glycolysis–driven metabolites sustain mitochondrial biogenesis and oxidative mitochondrial metabolism of cancer cells.10,12,42

The metabolic crosstalk in tumor microenvironment between CAFs and tumor cells that sustain tumor growth. In a dynamic tumor microenvironment, tumor and surrounding cells act as a functional network exchanging metabolic intermediates, oxygen, and nutrients that are essential for the survival, growth, and proliferation of the tumor.43–46 During metabolic reprogramming of NF to CAFs, a decrease in the expression of mitochondrial transcription factor A (TFAM) induces a loss of Cav-1 and a decline in oxidative phosphorylation (OXPHOS). 17,23 Mitochondrial biogenesis drives cancer aggressiveness, and in contrast to the observations made in cancer cells, during fibroblast transformation, the activity of complex I, complex II, and complex IV of OXPHOS decreases in concomitance with an increase in glycolysis.47–51 In glycolytic CAFs, an increase in MCT4 levels allows lactate export, followed by their influx in cancer cells via MCT1 that enters in TCA cycle driving oxidative metabolism—“reverse Warburg effect.”10,52,53 MCT4 expression also occurs in anaerobic cancer cells coupled to well-oxygenated cancer cells with OXPHOS activity. 53 Moreover, to sustain metabolic requirements for cancer proliferation, CAFs use other carbon sources to produce amino acids (e.g. glutamine and alanine), acting as suppliers of tumor cells.54,55
In prostate cancer, the close contact of fibroblasts with cancer cells triggers CAF metabolic reprogramming, with higher expression of glucose transporter isoform 1 (GLUT1), lactate production and export through de novo expressed monocarboxylate transporter 4 (MCT4). Interestingly, the metabolic modulation observed in prostate cancer cells was the opposite, since they decrease GLUT1 expression and increase lactate influx via monocarboxylate transporter 1 (MCT1). 56 This differential expression of MCT4 and MCT1 in CAFs and cancer cells has been associated with poor clinical outcome of prostate cancer. 57 In CAFs under co-culture conditions, MCT4 overexpression protects from death fibroblasts and breast cancer cells. 58 Recently, Shan et al. 59 reported that in co-culture systems, inhibiting MCT1 in CAFs results in a decrease in pyruvate kinase 2 expression, glucose import, and lactate production, with a concomitant decrease in pancreatic cancer cell migration. Moreover, in breast cancer CAFs, the activation of G-protein coupled estrogen receptor (GPER)/cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA)/cAMP response element–binding (CREB) and phosphoinositide 3-kinase (PI3K)/AKT/mechanistic target of rapamycin complex 1 (mTORC1) signaling pathways triggers the aerobic glycolysis switch, resulting in the secretion of pyruvate and lactate that fuels and increases the mitochondrial activity in cancer cells. 60
In proliferative cancer cells, the glutamine availability is essential for mitochondrial metabolism. Glutamine catabolism, more important than glucose metabolism in cancer cells, provides the anaplerotic carbons and oxoloacetic acid to fuel the TCA cycle, resulting in ATP and carbon skeleton production, essential for macromolecules and nucleotide synthesis.61–63 Glutamine metabolism in CAFs is able to stimulate proliferation in cancer cells.54,64 Interestingly, many studies associated the increase in glutaminolysis with carcinogenesis and showed that targeting this pathway could impair tumor proliferation.63,65,66 In fact, glutamine transporter SLC6A14 is often increased in cancer cells in the presence of CAFs in a glutamine concentration–dependent pattern. 8 Glutaminolysis also decreases cell stress through glutathione (GSH) production, releasing alanine and ammonia as catabolic-driven products.64,67,68
Yang et al. 55 reported that in an ovarian carcinoma model, CAFs upregulate glutamine anabolic pathway. The authors propose that co-targeting glutamine synthetase in CAFs and glutaminase in cancer could be an adjuvant strategy to the conventional ovarian cancer therapy.
In cancer microenvironment, it remains unclear if the only source of lipids is adipocytes. In melanoma and prostate cancer, fibroblasts transfer lipids to neighboring cells using microvesicles. 69 In pancreatic cancer, CAFs directly transfer lysophospholipids (lsyo-PLs) to cancer cells via lipid droplets. Recently, reinforcing the relevance of the metabolic synergy between CAFs and cancer cells, Lopes-Coelho et al. 70 demonstrated that CAFs cooperate with breast cancer cells as fatty acid suppliers in vitro and in vivo. Breast cancer cells exposed to CAFs-conditioned media increase their lipid cellular content and increase the expression of fatty acid transporter 1 (FATP1), favoring fatty acid uptake. Moreover, FATP1 is pointed as a putative target to disrupt lipid symbiosis between CAFs and breast cancer cells, thereby suppressing tumor progression.
Autophagy—a cellular phenomenon underlying CAF contribution for cancer progression
During metabolic reprogramming of CAFs, autophagy was also described as one of the upregulated pathways, being involved in CAF phenotype as active suppliers of energy sources for cancer cells. 2 Autophagy is a primarily survival phenomenon in which cells degrade their own components to obtain enough energy to keep themselves alive; but a prolonged autophagic process will induce cell death. In head and neck squamous cell carcinoma, it has been described that the inhibition in CAFs of phosphatidylinositol 3-kinase (Vps34), an upstream kinase of the autophagy pathway, decreases cancer progression. 71 In triple-negative breast carcinoma (TNBC), autophagic CAFs improve cancer cell features (e.g. proliferation and invasion) and EMT process through Wnt/β-catenin pathway. 72
Autophagic CAFs act as active players favoring the pro-tumorigenic nutrient-rich microenvironment. Cancer cells, in nutrient deprivation or energy restriction, can be fed by products resulting from autophagic CAFs. The induction of autophagy in CAFs is driven by the activation of TGFβ/SMAD signaling pathway,73–75 the mitochondrial fission factor (MFF) through NFkB signaling pathway, 76 and by the upregulation of mTOR/AMP-activated protein kinase (AMPK) pathway, 77 releasing lactate and glutamine. In contrast, mTORC1/c-Myc axis seems to be involved in the inhibition of the metabolic reprogramming of CAFs. 37 So, within a tumor, autophagy works in CAFs similar to glycolysis in cancer cells. In cancer cells, most of the times, autophagy and glycolysis do not work together.1,2 Actually, mTORC1 is involved in glycolysis impairment in cancer cells through hexokinase 2 inhibition, which activates autophagy as in this molecular process mTORC1 becomes inactive. 3 Nevertheless, some studies have shown that in particular genetic background, such as mutations in tumor suppressor gene p53 or oncogene KRas, glycolysis and autophagy can work simultaneously contributing for cancer cell survival, cancer progression, and metastasis.78,79
In breast cancer, CAFs with high levels of the lipid modified form of microtubule-associated proteins 1A/1B light chain 3B (LC3II), an autophagosome marker, are associated with a more aggressive cancer phenotype due to the release of high-mobility group protein 1 (HMG-1) by CAFs that binds to toll-like receptor 4 (TLR4) in breast cancer cells, enhancing their tumorigenicity. 80 Similarly, Lee et al. 81 described that pancreatic cancer cells are able to secrete transglutaminase (TG2) in order to stimulate fibroblasts to produce and secrete laminin A1, promoting tumor growth and protecting from cytotoxicity induced by drugs. Moreover, pancreatic ductal adenocarcinoma metabolism relies, in part, in alanine bioavailability secreted by autophagic stellate cells. Alanine acts as an alternative carbon source to fuel Krebs cycle, promoting tumor growth. 82
As mentioned above, glutaminolysis releases ammonia, which is toxic in normal cells, but in cancer cells ammonia is used to induce autophagy and neutralize internal pH.64,67,68 Furthermore, glutamine has also been implicated in the induction of CAF autophagy and consequently the promotion of cancer cell proliferation, which becomes protected from drug-induced cell death. 8 In addition, glutaminolysis and mTORC1 form a positive loop of mutual activation, which ultimately contributes for autophagy inhibition, 83 promoting cell growth and cancer progression.
Autophagy can regulate lipid metabolism by lipophagy.75,84 However, in CAFs, the possible parallelism between an increase in autophagy and in lipid metabolism is not completely explored so far. However, the transfer of Lsyo-PLs mediates the metabolic reprogramming of CAFs, inducing autophagy to support the metabolic requirements of cancer cells. 85
Adipocytes
Nowadays, it is already well established that adipose tissue is a complex organ with endocrine, metabolic, and immune roles. Adipose tissue comprises three main tissues: the brown adipose tissue that controls mainly thermogenesis, the mammary adipose tissue, and the white adipose tissue which is considered the most complex at the cellular level and responsible for the control of weight regulation.86–90
Adipose tissue has a role in cancer promotion and progression
Adipocyte-secreted factors (e.g. adipokines and adipocytokines; Figure 3) act on cancer cell proliferation, local invasion, metastatic spread, and resistance to chemotherapy and radiotherapy. 108 Breast cancer is one of the best examples of adipocytes–tumor cell interaction in which adipocytes act as fatty acid providers.93,97 Besides this, tumor cells express receptors for adipokines; for instance, leptin has been described as promoting proliferation in vitro in human breast cancer cell lines,109,110 and insulin growth factor 1 (IGF-1) secreted by adipocytes also stimulates breast cancer cell lines’ growth. 111 On the contrary, lower levels of adiponectin are associated with carcinogenesis and are linked to stimulation of apoptosis. 112

The role of adipocytes’ secretome in cancer proliferation, progression, and metastasis. Correlation between increased adipose tissues (ATs) with worse cancer patient’s prognosis is already established.91,92 Adipocytes, the major component of AT, are in a close contact with cancer cells in many solid tumors (e.g. breast cancer), and the relationship between adipocyte-secreted molecules in cancer progression is now recognized since adipocyte-secreted molecules can promote cancer cell proliferation, angiogenesis, and invasion.93,94 Signaling pathways related to carcinogenesis could be regulated by the adipokines secreted by adjacent adipocytes and recognized by specific receptors in tumor cell surface, in fact over 600 adipokines have now been identified.95,96 For instance, PI3K/AKT, MAPK/ERK and STAT3 signaling pathways could be activated selectively by leptin, hepatocyte growth factor (HGF), insulin-like growth factor 1 (IGF-1), and resistin, promoting cell survival and proliferation.97–101 Leptin, HGF, and VEGF-A produced by adipocytes had also been described as implicated in angiogenesis.102–104 Moreover, leptin has been described as exerting a pro-proliferative effect on cancer cells, while adiponectin acts as a pro-apoptotic factor. The remodeling of the extracellular matrix of the surrounding adjacent tumor area could also be associated with adipocytes since they release collagen VI and MMP11 promoting cancer invasion.105–107
In many cancer contexts, lipid metabolism has been already identified as determinant and as a major contributing factor for metabolic fitness of cancer cells. As mentioned above, cancer cells uptake metabolic substrates as fatty acids to support the rapid growth and provide energy necessary for biomass synthesis, migration, and invasion. 43 In ovarian cancer cell lines, adipocytes promote homing, migration, and invasion, 113 while in breast cancer cell lines, lipids exert growth-promoting effect and enhance tumor cell features (e.g. migration and invasion).4,97,111,114–120 In prostate cancer, a functional relationship between bone marrow adiposity and metastasis has been also established. 121
At the molecular level, fatty acids appear as one of the major contributors to the acetyl-CoA pool used in TCA cycle and support oxidative phosphorylation and consequently tumor growth. 122 Balaban et al. 4 reported that human primary adipocytes and mouse 3T3-derived adipocytes provide substrates to support the metabolic needs of breast cancer cell lines, concomitant with increased fatty acid storage and oxidation (β-oxidation). For instance, the metastatic potential of human oral cancer cells is linked to higher levels of fatty acid transporter (CD36) and increased expression of genes involved in β-oxidation, lipid biosynthesis, and intracellular lipid storage. 122 In breast cancer, it occurs due to an increase in gene expression of a rate-limiting enzyme (carnitine palmitoyltransferase 1 (CPT1)) in β-oxidation.4,123 Cancer cells adapted to hypoxic and acidic environment present an increase in lipoprotein uptake, accumulated as lipid droplets, and β-oxidation-derived acetyl-CoA to fuel TCA.123–127 It is noteworthy that inhibition of fatty acid uptake and/or metabolic-related pathways has gained attention currently as a putative adjuvant therapy to treat cancer.
CAAs play a role in cancer metabolic fitness
As previously stated, adipocytes are in a close contact with cancer cells in many solid and hematological tumors, and not only adipocytes are able to modulate and modify cancer cell behavior but also reciprocal effects of cancer cells on adipocytes have been reported

In cancer, the crosstalk between adipocytes, cancer cells, and cancer-associated adipocytes (CAAs) encourages tumor growth and metastasis. The close contact between tumor cells with adipocytes favor tumor progression through the supply of metabolic fuels and/or signaling mediators, driving the proliferation and invasion of tumor cells through fatty acid (FA) accumulation and fatty acids-related pathway activation.43,93,97,128 During carcinogenesis, adjacent adipocytes, CAA, undergo de-differentiation and exhibit delipidation, acquiring a fibroblast-like morphology concomitant with increased cancer cell aggressiveness.129,130 Adipocytes and CAA act as active players in the metabolic dynamic interplay between cancer and stromal cells through the production of hormones, growth factors, cytokines, adipokines, and metabolic substrates (fatty acids (FA)).97,105,109,128,131 A better knowledge of the cellular and molecular mechanisms involved in the crosstalk between adipocytes, cancer cells, and CAA may provide new insights and putative targets for the improvement of therapeutic strategies in cancer treatment.
In vitro and in vivo data suggest that during CAAs’ morphological changes, adipocytes decrease in cell number and size due to loss of their lipid content, exhibit fibroblast-like morphology, and have an altered secretome, inducing pro-tumor microenvironment remodeling and tumor progression.97,129,132 CAAs display enhanced secretion of fibronectin and collagen I, increased migratory/invasive abilities, and increased expression of the CAF marker FSP-1. In breast cancer, this multi-step process is dependent on the activation of Wnt/β-catenin pathway in response to Wnt3A secreted by cancer cells. 129 The induction of adipocyte de-differentiation by cancer cells is related to an increase in gene expression of adipokines and adipocytokines (tumor necrosis factor (TNF), IL-6, and IL1B) and genes involved in the remodeling of ECM genes (CCL2, CCL5, LEP, MMP11, and SERPINE1).97,133–135 Desmoplasia, which is the deposition of a fibrotic tissue surrounding cancer cells and is quite frequent in breast cancer, 136 can also be sustained by this de-differentiation process of adipocytes, resembling fibrotic tissue at the morphological and molecular levels. Picon-Ruiz et al. 134 reported a model in which cancer cell invasion into local fat establishes feed-forward loops to activate Src oncogene and maintain pro-inflammatory cytokine production favoring tumor-initiating cell abundance and metastatic progression.
This inflammatory environment can be responsible for the cachexia (loss of muscle and adipose tissues) observed in cancer patients. 137 However, more studies to clarify key molecular players in the crosstalk of adipocytes and cancer cells are needed.
Strengthening the importance of lipid metabolism in cancer context, several studies had proved that cancer cells itself are able to synthesize and metabolize lipids to support their growth and metastatic fate. For instance, the grade of breast cancer and poor prognosis has been correlated to increased levels of stearoyl-CoA desaturase (SCD) expression, a final key enzyme in newly synthesized monounsaturated fatty acids. 138 Also, a variety of studies linked the activity of fatty acid synthetase (FASN) with cancer progression. FASN inhibition induces endoplasmic reticulum stress in cancer cells enhancing cell death; in breast and prostate cancer, targeting FASN effectively suppresses growth and induces apoptosis in vitro and in vivo.139–141 FASN overexpression was also linked to drug resistance in breast cancer cells 142 and more recently in a group of breast cancer patients has been proposed as putative beneficiaries of a therapy with FASN inhibitors. 143
Adipocytes can be an active player in cancer progression and resistance to conventional treatment
The mechanisms underlying the obesity–cancer link are not fully understood. Recent evidences had shown the relevance of adipose tissue in cancer progression and outcome.91,92 For instance, in breast cancer scenario, it demonstrated, independent of a menopausal status, a correlation between lower survival and obesity. 144 In leukemia, poorer survival was described for obese patients comparing to patients within a normal range of weight.145,146
In the literature, most of the in vitro and in vivo studies do not consider the relevance of obesity in adipocyte relationship with cancer cells. Balaban et al. 4 reported that breast cancer cell lines co-cultured with adipocytes derived from obese women had an increased growth rate and migration properties compared to the effects observed in co-culture with adipocytes from normal individuals.
Adipocytes derived from obese individuals release two-fold higher IGF-1 levels than adipocytes from thin individuals; this may contribute to cancer growth in obese individuals since proliferation is enhanced in breast cancer cells exposed to obese adipocyte-conditioned media. 4 Moreover, Santander et al. 119 demonstrated in vivo that mammary tumors from obese mice had higher tumor volume than tumors from skinny mice.
Clinically, obese women display poor prognosis due to an increased resistance to conventional chemotherapy, but the mechanisms of resistance are not well known. Lehrmann et al. 147 demonstrated that in breast cancer cell lines, the sensitivity to docetaxel in vitro was decreased in the presence of serum from obese patients. In vivo models of leukemia also showed that adipose tissue niches permit a metabolic adaptation of cancer cells allowing chemotherapy evasion. 1 New knowledge about the epidemiological, clinical, and molecular data of obese patients is needed for a better understanding of the mechanisms underlying chemoresistance in these patients.
Monocytes/macrophages
Monocytes and macrophages belong to the myeloid lineage of leukocytes. Macrophages result from the differentiation, in tissues, of extravasating monocytes and undergo specific differentiation according to the local tissue microenvironment.148–150 These cells are mostly known by playing a critical role in injury emergence and resolution of infection. Also, they are involved in the maintenance of tissue homeostasis through remodeling and repair, and they are characterized by the ability to engulf invading pathogens, dying/dead cells, or cell debris. They also secrete a wide array of immunomodulatory cytokines as well as present antigens.148,149,151
Macrophages undergo specific differentiation according to the local tissue. Two extreme stages of polarization have been described, M1 and M2. M1 macrophages are considered as potent producers of pro-inflammatory cytokines and killers of microorganisms and tumor cells, whereas M2 cells are more prone to scavenging debris and promoting angiogenesis, tissue remodeling, and repair.152,153
TAMs have a role in cancer promotion and progression
TAMs seem to have acquired features shared by M2 macrophages

Tumor-associated macrophages (TAMs) are present as tumor aiders in all traits of cancer: initial oncogenic events, tumor growth, vascularization, migration, and metastasis. (a) Immunoediting and inflammation seem to mark the initiation of cancer. A malignancy-oriented microenvironment appears to contribute to TAMs “re-education”154,155 and includes the secretion, by tumor cells, of a cytokine mix that includes TGF-β1, acting here as a strong immune suppressor,156–158 and CSF-1, which blocks the maturation of monocytes into dendritic cells. 159 TAMs themselves become able to suppress other immune cells intervention by releasing, namely, IL-10 160 and arginase I.161–163 Similarly, an inflammatory milieu is always present in the initiation of the tumor. This microenvironment is achieved by the secretion by TAMs of reactive oxygen (ROS) and nitrogen species (NOS) that will generate mutations154,164,165 and also some cytokines associated to mutagenic events, including TNF-α and macrophage migration inhibitory factor (MIF). 166 (b) For the further events, inducers of a metastatic behavior in the tumor, a paracrine interaction between monocytes/macrophages and cancer cells is essential. One of those mechanisms involves the release of CSF-1 by tumor cells, which attracts monocytes into the tumor, which, in turn, secretes EGF, whose signaling promotes tumor cells migration.167–169 TAMs-derived TNF-α is another booster of the metastatic behavior as inducing MIF and extracellular matrix metalloproteinase inducer (EMMRIPN). This milieu still includes cancer cells–derived IL-4, CXCL12, FGF, HG, PDGF and TAMs-derived TGF-β,149,167,170 MMPs (MMP-2, MMP-7, MMP-9, and MMP-19), urokinase-type plasminogen activator (uPA), and IL-6, which act on ECM proteolysis, facilitating, thus, tumor cell migration and escape and additionally the release of eventual important mediators of tumor spreading.152,167,171
Hematopoietic cells are recruited to most solid tumors, and TAMs can abundantly populate the tumor mass.154,155 Strikingly, the vast majority of the studies on the subject suggest a causal relationship between macrophage high density and poor disease prognosis.174–176
TAMs’ influence on cancer is present in all traits of carcinogenesis (Figure 5). These cells participate in tumor initiation, growth, vascularization, and metastasis. Each trait seems to be affected by a particular subpopulation, displaying a more suitable phenotype for each function and recruited to strategic regions of the neoplasm, according to the chemokine expression pattern in microenvironment. 173
Chemokine (C-C motif) ligand 2 (CCL2)/C-C motif receptor 2 (CCR2) is the main determinant of monocyte recruitment into tumors. CCL2 (also known as monocyte chemoattractant protein (MCP)-1) expression has been positively associated with TAM accumulation in a wide panel of malignancies, including breast, ovarian, lung, glial cancers, and leukemia.173,177–180 Indeed, the induction of CCL2 expression in melanoma has resulted in more accumulation of TAMs. 181 In accordance, CCL2/CCR2 axis interruption has resulted in a reduced recruitment of monocytes/macrophages and a consequent decline in tumor burden.182,183
As mentioned, colony stimulating factor-1 (CSF-1) is the major lineage regulator for macrophages.184,185 This cytokine, whose effect is mediated by CSF-1 tyrosine kinase receptor (CSF-1R), is overexpressed by tumor cells in several neoplastic contexts, including breast, ovarian, prostate, and colorectal cancer,185–190 and has been associated with TAM accumulation, more aggressive malignancies, and consequent poor prognosis. The ablation of CSF-1/CSF-1R interaction, either by genetic or therapeutic approaches, has resulted in the reduction of monocyte/macrophage recruitment or function and subsequent impairment of tumor growth and spread. On the contrary, the induction of CSF-1 overexpression has been shown to accelerate tumor progression. 187 TAMs have still been identified as contributors to tumor progression by inducing COX-2 as a consequence of IL1β-mediated stimulation of ROS-Src-MAPK signaling. 191
TAMs—metabolic adjustment in cancer
The role of TAMs in the metabolic adjustment of tumor cells themselves, so far, seems to be mostly related to the angiogenic switch: as a tumor becomes larger, its metabolic demands increase, which requires a more developed vascular structure.192–194 This transition is simulated by TAMs, which respond to hypoxia and promote vascularization through several mechanisms.
Although little is known about the metabolic reprogramming of monocytes/macrophages in the context of carcinogenesis (Figure 6), this is another feature underlying their role as cancer “helpers.” 195 TAMs seem to show, generally, increased aerobic glycolysis 196 via Akt/mTOR and HIF-1α stabilization.197–199 An enhanced fatty acid biosynthesis and uptake is another metabolic adjustment that may explain an enhanced pro-inflammatory, pro-tumorigenic profile of TAMs.197,200,201 An increase in the expression of genes involved in glutamate transport and metabolism was observed in TAMs isolated from tumors. 202 In addition, the overexpression of arginase 1 (ARG 1) in TAMs seems to be associated to an enhancement of tumor cell proliferation, 203 probably via ARG1-dependent polyamine synthesis pathway, while suppressing tumor cytotoxicity by reducing nitric oxide production.203,204
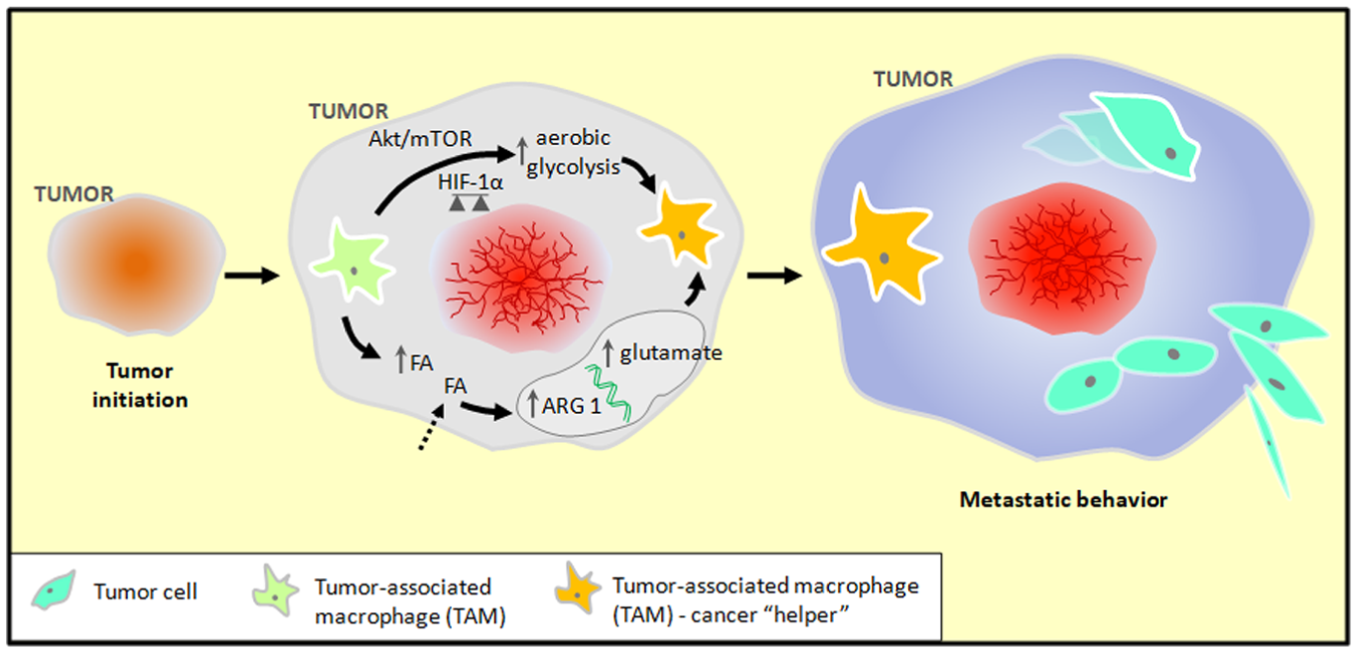
Tumor-associated macrophage (TAM) metabolic remodeling in cancer. TAMs, in general, show increased aerobic glycolysis 196 via Akt/mTOR and HIF-1α stabilization,197–199 enhanced FA biosynthesis and uptake,197,200,201 and overexpression of genes involved in glutamate transport and metabolism 202 and arginase 1 (ARG 1).203,204 These metabolic features may also explain the pro-inflammatory and pro-tumorigenic profile of TAMs.
Learning more about the interaction between TAMs and cancer metabolism should be a challenge for future research.
Highlights
Cancer survival and progression are not exclusively dependent on the cancer cells. In fact, carcinogenesis depends on the contribution of several cellular and molecular components of the tumor microenvironment, being non-cancerous stromal cells crucial peons.
Stromal non-cancerous cells are pivotal players in the maintenance of the network that sustains and feeds cancer cells. This network is composed of metabolic and signaling pathways needed to accomplish the high demanding of energy and biomass, undoubtedly essential for cancer initiation and progression. The destabilization of this equilibrium can be a suitable strategy to define new and powerful therapeutic approaches. The effective abrogation of the metabolic crosstalk is for sure a challenging field of cancer research.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported through iNOVA4Health—UID/Multi/04462/2013, a program financially supported by Fundação para a Ciência e Tecnologia (FCT; PhD ProRegeM program, PD/BD/128337/2017)/Ministério da Educação e Ciência through national funds and co-funded by FEDER under the PT2020 Partnership Agreement.