
Abstract
Transient receptor potential melastatin 8 (TRPM8), a membrane ion channel, is activated by thermal and chemical stimuli. In pancreatic ductal adenocarcinoma, TRPM8 is required for cell migration, proliferation, and senescence and is associated with tumor size and pancreatic ductal adenocarcinoma stages. Although the underlying mechanisms of these processes have yet to be described, this cation-permeable channel has been proposed as an oncological target. In this study, the glycosylation status of the TRPM8 channel was shown to affect cell proliferation, cell migration, and calcium uptake. TRPM8 expressed in the membrane of the Panc-1 pancreatic tumoral cell line is non-glycosylated, whereas human embryonic kidney cells transfected with human TRPM8 overexpress a glycosylated protein. Moreover, our data suggest that Ca2+ uptake is modulated by the glycosylation status of the protein, thus affecting cell proliferation.
Keywords
Introduction
Transient receptor potential (TRP) represents a super family of ion channels expressed in all animal kingdom, where they take part in many cellular functions.1–5 The family is sub-divided into seven major subfamilies: TRPC (canonical), TRPV (vanilloid), TRPA (ankyrin), TRPM (melastatin), TRPP (polycystin), TRPML (mucolipin), and TRPN (also known as NOMPC (no mechanoreceptor potential C)). TRP proteins are mostly unselective channels for monovalent and divalent cations and are gated by numerous stimuli such as temperature, chemical substances, mechanical and osmotic stresses, or signalling molecules, therefore acting as receptors in diverse sensory processes. In the last years, TRP ion channels are recognized also as important key proteins in the processes of cell proliferation, migration, adhesion, or epithelial-to-mesenchymal transition, through changes in their expression levels or activation. Among all the family members, special attention was given to studies of transient receptor potential melastatin 8 (TRPM8) in the tumorigenesis processes. The fact that the protein was first cloned from the human prostate recommended TRPM8 as a sensor for different other stimuli except from cold, as it was mostly studied until then. Later on, numerous studies brought information about the increased expression of the channel in breast cancer, prostate cancer, and lung cancer and its implication in tumorigenesis processes. 6 The complexity of TRPM8 function might be the result of the possibility to form various multimeric complexes which are expressed in plasma membrane or intracellular organelles. It is also established that posttranslational modifications including phosphorylation, cysteine modification, and glycosylation modulate channel trafficking, gating, temperature perception, or oligomerization (reviewed in Voolstra and Huber 7 ). Special attention was given to N-linked glycosylation state of TRPM8, which is involved in assembling functional ion channels and participates to subunit trafficking. 8 Stages of N-linked glycosylation that occur at specific Asn residues can be experimentally assessed using their sensitivity to endoglycosidases. For instance, N-glycosidase F releases the intact oligosaccharide, whereas endoglycosidase-H cleaves the “high-mannose” oligosaccharides, making possible to disparate weights of ‘mature’ and ‘immature’ glycoprotein. 9 A direct experimental approach that maintains the viability of cells is the use of low concentrations of tunicamycin, an antibiotic that prevents glycosylation of proteins through inhibition of N-acetylglucosaminylpyrophosphorylpolyisoprenol synthesis. 10 Different studies have shown that TRPM8 exists in two glycosylation states, which vary depending on the cellular context where this post-translational modification occurs, affecting the function of the channel.10,11 Channels challenged with tunicamycin were expressed on the cell surface, but they displayed lower current amplitudes and a shift in the potential half-maximal activation (V1/2) toward positive values and lower temperature sensitivities. 10
However, the studies published thus far are limited to experiments performed in channels heterologously expressed in HEK cells, circumventing the variability of TRPM8 expression in native tissues. Here, we present a study performed on the endogenous TRPM8 expressed in cell lines originated from pancreatic ductal adenocarcinoma (PDAC). TRPM8 expression is increased in PDAC cells and patients compared to that of pancreatic ductal cells derived from non-tumoral tissues. 5 There is no consistent difference in surface expression between cells heterologously expressing the channel and cancer cells; however, the biophysical properties differ, and cancer cells display whole-cell currents with a lower amplitude and menthol sensitivity. 3
In this context, the study presented here was designed (1) to identify any differences in TRPM8 glycosylation between native (as is the case for the Panc-1 cell line) and heterologously (HEK/M8) expressing cells and (2) to determine whether these differences are linked to carcinogenesis.
Results
TRPM8 is non-glycosylated in PDAC cells
Previous studies performed on dorsal root ganglia showed that the native TRPM8 is highly glycosylated in vivo, which affects protein trafficking and functional properties of the channel.8,12 In our previous study, we showed that TRPM8 is expressed to a variable degree in PDAC cells and we assumed that the channel is in an un-glycosylated form. To determine whether this is the case, we examined protein expression in four different PDAC cell lines as compared to HEK-293 cell line, which expresses the heterologous channel. The cells were treated with 10 μg/mL of tunicamycin (an N-glycosylation inhibitor) for 24 h. Alternatively, cell homogenates were treated with the deglycosylating enzymes endoglycosidase H

TRPM8 expression and glycosylation level in four PDAC cell lines and TRPM8-transfected HEK cells. Cells were grown in their specific media, harvested, and lysed in 1% Triton X-100 lysis buffer. Equal amounts from the total lysate were incubated overnight with the deglycosylating enzymes EndoH (E) or PNGase (P). Digested proteins were separated in SDS-PAGE, transferred on nitrocellulose membrane, and probed with anti-TRPM8 polyclonal antibodies and calnexin (CNX).
TRPM8 currents have different kinetics in native and heterologous systems
The sensitivity to specific TRPM8 agonists highly depend on this glycosylation state,8,12 and experiments on HEK-expressing cells showed that TRPM8 channels expressed in heterologous systems display different biophysical properties as compared with channels in native systems. 10
Therefore, we monitored responses to menthol with the patch-clamp method in Panc-1 and HEK/M8 cell lines. The protocol used is shown in the Figure 2(a) inset. Cells were held at −60 mV, and voltage ramps from −100 to +100 mV were applied in the presence and absence of 300 μM menthol. The current density was measured at +100 mV; it was 59.8 ± 16.2 pA/pF in Panc-1 cells and 91.3 ± 14.5 pA/pF in HEK/M8 cells (Figure 2(a) and (b)). Notably, the current densities measured in these experiments were significantly different (p = 0.04, n = 5). More importantly, Panc-1 cells showed V1/2 values of 85.7 ± 7.2 mV, significantly more positive than the 66.6 ± 5.3 mV measured in HEK/M8 cells (p = 0.04; Figure 2(c)). In conclusion, the lack of glycosylation has an important role in the biophysical functions of TRPM8.

Panc-1 cells exhibited a shift in the voltage activation of TRPM8 towards more positive potentials. (a) Current–voltage relationships of HEK/M8 cells recorded in control conditions (CTRL) in the presence of 300 μM menthol at RT and 1 min after menthol washout (wash-out). Continuous lines represent averages of n = 6 cells ± SEM (grey lines). The applied protocol is described in the figure inset. (b). Current–voltage relationships (IV) of Panc-1 cells recorded in the same conditions as described in panel A. (c). Column graphs showing averages ±SEM (n = 6) of V1/2 activation for HEK/M8 cells (grey columns) versus Panc-1 cells (black columns). Results are significantly different (p = 0.04).
Additionally, Ca2+ microfluorimetry was used to monitor channel function by comparing the responses elicited by menthol in HEK/M8 and Panc-1 cells. The protocol consisted of three menthol pulses of 1 min each separated by 4 min of Ringer washout. Consistent with a previous study, 3 in HEK/M8 cells, calcium transients evoked by menthol elicited responses with comparable amplitudes (measured as ΔF/F0) after each of the three pulses of approximately 0.4 (Figure 3(a)). In Panc-1 cells, the responses to menthol after each menthol application were comparable for the first two pulses, though a significantly lower value of 0.2 ± 0.02 was obtained for the third pulse, as shown in Figure 3(b) (n = 49 HEK/M8 cells, n = 24 Panc-1 cells). Moreover, in HEK/M8 cells, the menthol responses were robust and unaltered after three consecutive applications (Figure 3(a)). In contrast, only approximately 63% of Panc-1 cells responded to the second pulse and 29% to the third. These results point toward different kinetics of calcium uptake in these two cell lines.

Calcium uptake through TRPM8 channels. (a) Representative experiments showing TRPM8 activation after three menthol pulses (Me1, Me2, and Me3) applied to HEK/M8 cells. (b). The same protocol described in A for Panc-1 cells. (c) Amplitude of [Ca2+]i in responding cells, represented as the difference between the peak value and the baseline value for menthol-induced responses. Each column represents the mean ± SEM of the response from n = 50 HEK/M8 cells and n = 24 Panc-1 cells. Only in the third pulse were the differences between Panc-1 cell responses versus HEK/M8 cells significant (***), whereas ns represents the insignificant responses for the first two pulses.
As previously reported, the non-glycosylated channels exhibited a lower sensitivity to specific agonists compared to that of the glycosylated form of the protein. 10 To determine whether these differences are reproducible with Ca2+ microfluorimetry, we exposed control untreated and tunicamycin-treated HEK/M8 cells to three 300 μM menthol pulses. As expected, the results indicated a significantly lower Ca2+ uptake, decreasing from 0.4 ± 0.01 to 0.22 ± 0.01 ΔF/F0 for non-glycosylated versus glycosylated channels (Figure 4(a)).

Channel deglycosylation inhibits [Ca2+]i uptake. (a) Fluorescence changes from baseline and ΔF normalized to F0 at the peak of the menthol response (***p < 0.001, n = 30) in untreated (CTRL) and tunicamycin-treated cells. (b) No significance in [Ca2+]i uptake in tunicamycin-treated Panc-1 cells versus control upon menthol stimulation (n = 49 without tunicamycin, n = 24 with tunicamycin).
Similarly, Panc-1 cells were exposed to 10 μg/mL tunicamycin for 24 h and challenged with three menthol pulses. In contrast to the results with HEK cells, no significant change was observed in [Ca2+]i uptake between treated and untreated cells (Figure 4(b)).
From these data, so far, it is clear that the glycosylation status of the cells is related to Ca2+ uptake and channel kinetics. However, the role of the channel in tumorigenesis has yet to be revealed. If glycosylation status of the channel is important in this process, we should be able to detect an influence on cell proliferation or cell migration.
TRPM8 inhibits PDAC cell proliferation
In our previous report, we showed that TRPM8 inhibits cell migration in Panc-1 cells. 3 To determine whether TRPM8 affects cell proliferation, we performed 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) tests in cells treated with small interfering RNA (siRNA)-TRPM8. Panc-1 cells transfected with TRPM8 siRNA showed a 20% higher proliferation rate than that of scrambled siRNA-transfected cells (Figure 5(a)). Observed also in studies of cell migration, 3 adhesion, or spheroid sprouting in vitro, 13 TRPM8 acts as an inhibitor of processes related to tumorigenesis, in this case proliferation of PDAC cells. Next, we examined whether tunicamycin interferes with the proliferation rate of cells. Therefore, we compared the proliferation rates in control and tunicamycin-treated Panc-1 cells. As shown in Figure 5(b), 10 μg/mL of tunicamycin had no effect on the proliferation of Panc-1 cells. Besides not affecting the channel, which is already de-glycosylated, tunicamycin did not interfere with other cellular process, at least in these experimental conditions, confirming results obtained in previous studies which demonstrated that Panc-1 cell proliferation is not affected in the presence of <2.0 mg% tunicamycin. 14 The efficacy of siRNA transfection was assessed through western blotting at the end of the MTS experiments (after 24 h) using calnexin as a reference (Figure 5(c)).

TRPM8 inhibits the proliferation rate of Panc-1. (a) At 24 h time-points, cell proliferation was significantly lower in Panc-1 cells compared to siRNA-treated cells. (b) Tunicamycin treatment for 24 h did not affect cell proliferation (***p < 0.001; ns indicates non-significant p values). (c) Western blot analysis of TRPM8 expression upon siRNA treatment in comparison to Calnexin (CLX) protein as a loading control.
Proliferation related to TRPM8 glycosylation
A better insight of the putative relationship between proliferation and glycosylation status should be assessed from studies on HEK/M8 cells. Indeed, experiments performed in the presence and in the absence of tunicamycin and in siRNA-transfected versus scrambled siRNA cells revealed some interesting information. The efficacy of siRNA transfection was assessed through western blotting at the end of the MTS experiments (after 24 h) using calnexin as a reference (Figure 6, bottom). We used only the experiments in which siRNA had a consistent effect. As illustrated in the left panel from Figure 6, tunicamycin had a strong significant effect (p < 0.005) on HEK/M8 proliferation. The contribution of TRPM8 protein to this effect is illustrated in the right panel. First, we notice that in the absence of the protein, cells have a lower proliferative rate, which implies that TRPM8 stimulates proliferation in HEK/M8, contrary to the results obtained in Panc-1 cells. Second, the effect of tunicamycin is not significant (p > 0.05) in cells that lack the TRPM8 protein. Concluding, we notice from these data that the glycosylation is important in cell proliferation and the un-glycosylated form may have a protective role in pancreatic adenocarcinoma.

Contribution of TRPM8 glycosylation to cell proliferation rate as determined by MTS assays. HEK/M8 cells were treated with 10 μg/mL tunicamycin for 24 h. Columns represent the mean ± SEM of triplicate experiments for each condition. At 24 h time points, cell proliferation was significantly lower in tunicamycin-treated cells compared to non-treated cells for scrambled siRNA condition. (a) Tunicamycin treatment for 24 h did not affect cell proliferation in cells where TRPM8 was silenced (***p < 0.001; ns indicates nonsignificant p values). (b) Western blot analysis of TRPM8 expression upon siRNA treatment in comparison to Calnexin (CLX) protein as a loading control.
We intended to assess also whether the glycosylation influences the migration of cells. Unfortunately, due to low proliferation rate after tunicamycin treatment, the experiments on HEK/M8 cells could not be performed. However, we show in Figure 7 that in Panc-1 cells, tunicamycin dramatically decreased migration rate. However, this effect cannot be related to TRPM8 channel because it can be noticed also in TRPM8-silenced cells.
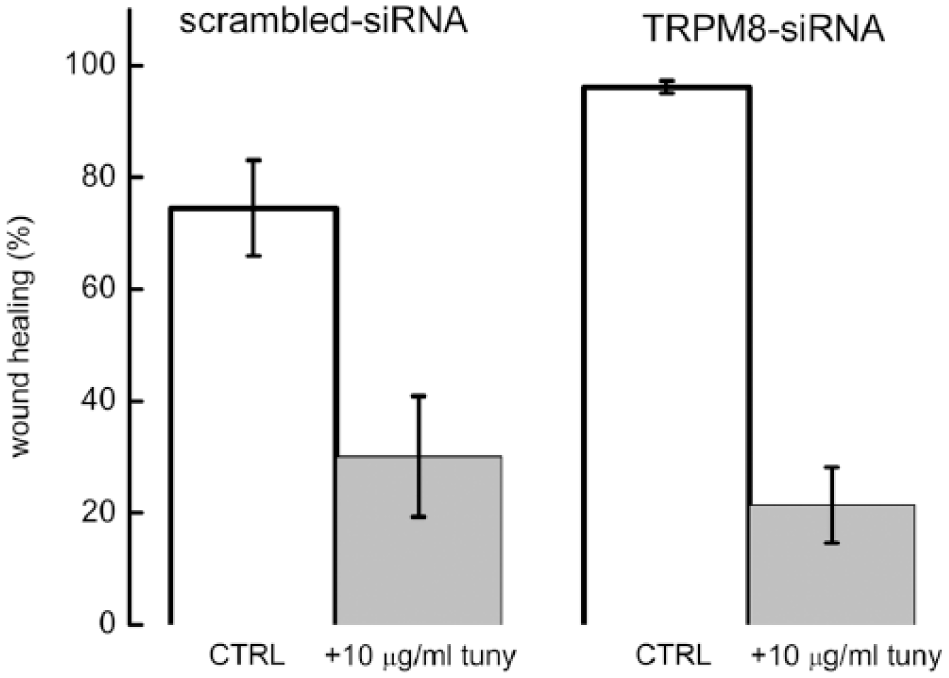
The effect of tunicamycin on the migration of Panc-1 cells. The experiments were performed in Petri dishes and wound-healing assay was carried out at 24 h after wound formation in control cells (treated with scrambled siRNA; left images) and cells treated with anti-TRPM8 siRNA (right images). The migration of Panc-1 was significantly decreased after tunicamycin treatment (experiments, n = 9; ***p < 0.001).
To determine whether tunicamycin can induce changes in the conductive state of Panc-1 cells, we analyzed menthol-evoked currents in the presence and absence of tunicamycin. Current amplitudes did not change (as shown in Figure 8) as well as the V1/2 values.

Menthol-induced currents are not significantly changed by tunicamycin. Statistical analysis of the whole-cell experiments at membrane potentials of −80 and +80 mV (n = 5 experiments with tunicamycin treatment; n = 9 experiments without tunicamycin).
Discussion
The aim of this study was to explore the differences between TRPM8 function natively expressed in pancreatic cancer cells and heterologously expressed in their counterparts. Although it is predominantly described as a temperature sensor in the peripheral nervous system, this channel has recently been shown to be functional in cells that are not necessarily sensitive to temperature changes and participate in migration, cell viability, and senescence.3,15
This study demonstrated that in Panc-1 cells, activation of TRPM8 channels produced uptake of Ca2+ as measured by menthol-induced currents and Ca2+ microfluorimetry. The menthol-activated currents were characterized by outward rectification and reversal of potentials close to 0 mV, as described for membrane TRPM8 currents. 16 From patch-clamp studies, activation of TRPM8 is defined by the potential of the half-maximal activation (V1/2). In sensory neurons, inhibitory agents shift the V1/2 values toward more positive potentials, 17 whereas cold and menthol negatively shifted this parameter. Interestingly, Panc-1 cells exhibited a right shift of V1/2 consistent with the results reported by Pertusa et al. 10 when the channels expressed in HEK cells were deglycosylated with tunicamycin.
These results suggest that TRPM8 has a different gating mechanism in the Panc-1 cell line. The study presented here is based on the reasoning that the difference between channel activation in HEK/M8 and Panc-1 cells is the result of channel deglycosylation. This hypothesis was confirmed by the treatment with enzymes that remove glycans. Endo H selectively removes N-linked glycans with high mannose and/or hybrid types from glycoproteins but does not remove the complex type. 18 PNGase F removes all N-linked glycans from glycoproteins. 19 The HEK/M8 cell lysates containing TRPM8 were sensitive to PNGase F and tunicamycin, exhibiting lower signal intensities, whereas it had no effect on Panc-1 cells. We conclude from these results that TRPM8 is non-glycosylated in Panc-1 cell, as well as in other three cell lines derived from pancreatic adenocarcinoma. The non-glycosylated form may explain the shift in V1/2 in Panc-1 cells, consistent with similar results obtained from non-glycosylated TRPM8 overexpressed in HEK cells. 10 Other studies showed that N-glycans of the K channel Kv3.1 influence conducting and non-conducting functions of the Kv3.1 channel. 20 Notably, the outward ionic currents of the non-glycosylated channel had slower activation and deactivation rates than those of the glycosylated Kv3.1 channel. Based on these previous reports, we hypothesized that Panc-1 TRPM8 is expressed in a non-glycosylated form that changes the conducting properties of the protein in PDAC cells.
We show here that TRPM8 has a molecular mass of approximately 138 kDa compared to Panc-1 cells that have a channel of approximately 130 kDa. These data are consistent with previous reports that found a shorter isoform in neuroblastoma cells 21 and prostate,22,23 which was predominantly expressed in the ER of the cells. We previously showed that this is not the case for Panc-1 cells, which express channels in their plasma membrane that are readily activated by menthol or icilin. 3 Interestingly, this channel inhibited the migration of PDAC cells, confirming the results obtained from PC-3 prostate adenocarcinoma cell lines; however, these results contrasted to those from studies on oral squamous carcinoma 24 and glioblastoma cells. 25 Most studies thus far have not elucidated the manner in which these features affect tumorigenesis. This report shows that Panc-1 cell proliferation rates are influenced by the presence of TRPM8, as demonstrated by experiments performed with siRNAs, which were extremely efficient and silenced the protein almost entirely. Cell proliferation increased by 30% in Panc-1 cells in the absence of the channel, while it strongly suppressed the proliferation in HEK/M8 cells.
Furthermore, the effect of glycosylation on proliferative status of cells was also addressed. Because low concentrations of tunicamycin are compatible with cell survival in Panc-1, we used this protocol to assess changes in cell proliferation upon deglycosylation. We hypothesized that tunicamycin treatment affected TRPM8 in HEK/M8 cells, whereas a minor or no effect would be observed in Panc-1 cells. Indeed, we found that the non-glycosylated form of the channel decreased cell proliferation rates by approximately 23% only in HEK/M8-expressing cells.
In conclusion, we propose the hypothesis that the capacity of TRPM8 to act as a calcium channel at the plasma membrane is modulated by N-glycosylation, which affects the proliferative capacity of the cell.
Numerous studies have shown that tunicamycin induces ER stress and influences the proliferation rates of cells. 26 Other previous reports indicated that experimental deglycosylation of TRPM8 induces loss of sensitivity to physical and chemical agonists. In our experiments, Ca2+ uptake as measured by Ca2+ microfluorimetry was significantly inhibited upon tunicamycin treatment of HEK/M8 cells and had no effect on Panc-1 cells. We concluded that the decreased proliferation is a result of TRPM8 deglycosylation because in the absence of the channel, HEK/M8 cell proliferation was not diminished, as shown in Figure 6. Further studies are needed to determine the signaling pathways involved in this process. A comparison with non-tumoral pancreatic epithelial cells is being undertaken in our lab.
In conclusion, these results showed that TRPM8 is nonglycosylated in Panc-1 cells. In this form, the protein has the ability to transport Ca2+ ions, although the mechanisms of ion transport are different. Additionally, non-glycosylated protein protects against high proliferation rates as it also impeded migration of PDAC cells.
Experimental procedures
Cell culture
Stably hTRPM8-transfected HEK293 cells (referred to herein as HEK/M8) were obtained from Professor Thomas Voets, Katholieke Universiteit Leuven, Belgium and maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin–streptomycin (100 IU/mL–0.1 mg/mL), and 500 μg/mL G418. PDAC pancreatic cells were cultured in DMEM supplemented with 10% FBS and penicillin–streptomycin (100 IU/mL–0.1 mg/mL). Panc-1, Mia-Paca 2, and BxPC3 were purchased from American Type Culture Collection (Rockville, MD, USA) and PK-9 was a gift from professor Akira Horii, Division of Molecular Pathology, Department of Pathology, Tohoku University Graduate School of Medicine, Sendai, Japan. All culture media and supplements were purchased from Invitrogen (Carlsbad, CA, USA).
Western blot assay
Total protein was obtained by lysing cells in Triton X-100 buffer (1% Triton X-100, 150 mM NaCl, 1.5 mM MgCl2, 1 mM ethylenediaminetetraacetic acid (EDTA)) or 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)–3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) buffer (2% CHAPS in 50 mM HEPES, 200 mM NaCl, pH 7.5; Sigma-Aldrich, Inc., St. Louis, MO, USA) supplemented with a protease inhibitor cocktail (Roche, Basel, Switzerland) for 30 min on ice. Equal amounts of protein (30 µg) were electrophoretically analyzed on a 7.5% polyacrylamide gel. The proteins were then transferred onto a nitrocellulose membrane (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). The membrane was blocked in 10% nonfat dry milk for 1 h at room temperature and then soaked for 1 h at room temperature either in a 1:500 diluted primary rabbit polyclonal anti-TRPM8 antibody solution (code: sc-130903; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) or in a 1:5000 diluted primary rabbit polyclonal anti-calnexin solution (code: ab22595; Abcam, Cambridge, UK), both of which were diluted in 1% milk in PBS–0.1% Tween 20. Proteins were detected with anti-rabbit IgG horseradish peroxidase–linked secondary antibodies (Santa Cruz Biotechnology, Inc.) and anti-mouse IgG and diluted in the same buffer as the primary antibody (1:10,000) for 1 h at room temperature (RT), followed by chemiluminescence detection using ECL Prime (GE Healthcare Life Sciences, NJ, USA) according to the manufacturer’s instructions.
RNA interference-mediated gene silencing
Panc-1 and HEK/M8 cells were grown to ~70% confluence in medium without antibiotics, trypsinized, and incubated for 48–72 h with fresh medium containing 300 nM of siRNA directed against human TRPM8 (sc-95009; Santa Cruz Biotechnology, Inc.) or 300 nM of scrambled control siRNA (sc-37007; Santa Cruz Biotechnology, Inc.), using Lipofectamine RNAiMAX (Invitrogen) as a carrier and incubated for 48–72 h. For confirmation of TRPM8 knockdown, the cells were harvested by trypsinization and lysed in HEPES-CHAPS buffer. Then, the total protein content was determined with a bicinchoninic acid (BCA) assay kit (Pierce, Rockford, IL, USA), and 30 µg of total protein was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by western blotting.
Electrophysiological measurements
Whole-cell patch-clamp recordings were performed with borosilicate glass pipettes (Warner Instrument, Hamden, CT, USA), which were heat-polished immediately before the experiment using a vertical puller (WPI, Berlin, Germany) to a resistance of 2–5 MΩ. Data were sampled with an AxoPatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA), low-pass filtered at 5 kHz and digitized at 10 kHz through a DigiData 1322A interface (Molecular Devices) driven by pCLAMP 8.1 software (Molecular Devices). Cells plated on Petri dishes were perfused with a standard bath solution at room temperature containing (in millimolar) 160-NaCl, 4.5-KCl, 2-CaCl2, 1-MgCl2, and 10-HEPES adjusted to pH 7.4 (360 mOSm/kg). Activated currents were measured in the presence of the indicated menthol concentrations.
Pipettes were filled with an intracellular solution containing (in millimolar) 140-CsCl, 5-EGTA (2 ethylene glycol-bis[-aminoethyl ether]-N,N,N,N-tetraacetic acid), and 10-HEPES adjusted to pH 7.4 (270 mOsm/kg). TRPM8 currents were induced by a depolarizing voltage ramp of 400 ms duration from −100 to +100 mV (slope 200 mV/s) from a holding potential of −60 mV and normalized to the cell membrane capacity (current density (pA/pF)).
To estimate the shifts in the voltage dependence of TRPM8 activation in HEK293 cells, current–voltage (I-V) relationships obtained from voltage ramps were determined with equation (1)

where g is the whole-cell conductance, Erev is the reversal potential of the current, V½ is the potential for half-maximal activation, and s is the slope factor. The assumption of a linear conductance is based on the observation that open TRPM8 channels display ohmic I-V dependence. 16 For each cell, fitting was started by analysis of a condition with strong channel activation, typically 300 μM menthol at 20°C. Erev was fixed at a value close to the measured reversal potential of the current evoked by menthol.
Intracellular Ca2+ imaging
Cells cultured on coverslips were incubated for 30 min at 37°C in standard extracellular solution (see solutions below) containing 2 μM Calcium Green-1 AM and 0.02% Pluronic F-127 (both from Invitrogen), incubated for loading for 30 min, and then left to recover for another 30 min before use. Coverslips were mounted in a Teflon chamber (RC-40HP; Harvard Apparatus, Holliston, MA, USA) on the stage of an Eclipse TE300 inverted microscope (Nikon, Tokyo, Japan) and left for 5 min to adapt to the extracellular solution flux at 25°C. Cells were illuminated with an Optoscan monochromator (Cairn Instruments, Faversham, UK), and the fluorescence changes were captured with a 12-bit CCD SensiCam camera (PCO, Kelheim, Germany). The data were recorded using Axon Imaging Workbench 4.0 (INDEC BioSystems, Mountain View, CA, USA). After background subtraction, data were quantified as ΔF/F0 for each recorded cell, representing the ratio between the maximum fluorescence change during the stimulus and the baseline fluorescence before the stimulus.
MTS tests
To perform a cell proliferation test, Panc-1 and HEK/M8 cells were grown under different conditions, silencing or inhibitor treatment, with proper controls. At the end of the treatment, the cells were incubated with MTS reagent (G3580; Promega, Madison, WI, USA) following the manufacturer’s instructions. The absorbance of the final product of the reaction was measured at 490 nm. Final values, obtained after background subtraction, were represented in GraphPad Prism.
Migration experiments
The migration experiments were performed using the scratch wound technique, as previously reported. 3 Briefly, cells with 100% confluence grown on 35-mm diameter Petri dishes (Nunc; Thermo Scientific, MA, USA) were scratched with a 200-μL pipette tip. For half of the cells, tunicamycin was added in the culture medium. The wound closure was monitored using a microscope at the time it was created and 24 h later. The wound area was measured with ImageJ software. The percentage of wound closure was measured from three Petri dishes for each condition at 0 and 24 h in the presence and in the absence of tunicamycin.
Statistical analysis
Data are expressed as the mean ± standard error of mean (SEM); n represents the number of experiments performed, and Student’s t test was used for statistical analysis.
Footnotes
Acknowledgements
R.U. and G.C. performed all the experiments with help from A.D. and L.S. V.R and D.F.M. analyzed the data. F.C. and D.C. drafted the manuscript. D.C. supervised the project. All authors reviewed the results and approved the final version of the manuscript. We acknowledge Dr Simona O. Dima from the Fundeni Clinical Institute for kindly offering PDAC cell lines. R.U. and G.C. contributed equally.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.