
Abstract
Immunotolerance is one of the hallmarks of malignant tumors. Tumor cells escape from host immune surveillance through various mechanisms resulting in tumor progression and therapeutic resistance. Interlukin-6 is a proinflammatory cytokine involved in many physiological and pathological processes by integrating with multiple intracellular signaling pathways. Aberrant expression of interlukin-6 is associated with the growth, metastasis, and chemotherapeutic resistance in a wide range of cancers. Interlukin-6 exerts immunosuppressive capacity mostly by stimulating the infiltrations of myeloid-derived suppressor cells, tumor-associated neutrophils, and cancer stem-like cells via Janus-activated kinase/signal transducer and activator of transcription 3 pathway in tumor microenvironment. On this foundation, blockage of interlukin-6 signal may provide potential approaches to novel therapies. In this review, we introduced interlukin-6 pathways and summarized molecular mechanisms related to interlukin-6-induced immunosuppression of tumor cell. We also concluded recent clinical studies targeting interlukin-6 as an immune-based therapeutic intervention in patients with cancer.
Introduction
Chronic inflammation contributes to the initiation and progression of various cancers.1,2 Immunosuppression may be a key mechanism involved in the inflammation-induced tumorigenesis by restraining immune surveillance.3,4 Adaptive immunity effectively protects the body from cancer progression and induces tumor dormancy.5–7 Nevertheless, by recruiting monocytes/macrophages, myeloid-derived suppressor cells (MDSCs), and tumor-associated neutrophils (TANs), activated innate and humoral immune responses cancel the adaptive immunity protection, followed by the generation of tumor growth–dependent stroma and inhibition of immunocompetence in cancer patients.8–11
Interlukin-6 (IL-6), known as a pleiotropic cytokine, is produced by multiple immune cells. Previous studies showed that aberrant expression of IL-6 detected in various cancers (e.g. breast cancer, lung cancer, liver cancer, colorectal cancer, prostate cancer, melanoma) in both messenger RNA (mRNA) and protein level was relevant to oncogenesis, tumor progression, multidrug resistance, and poor clinical outcomes in vitro and in vivo.12–15 In clinical specimens, elevated IL-6 also has been associated with advanced stage and poor prognosis of patients with cancer. Mechanically, IL-6 regulates the growth and progression of malignancies by inhibiting apoptosis, stimulating angiogenesis, and inducing drug resistance via several signaling pathways, especially the Janus-activated kinase/signal transducer and activator of transcription 3 (JAK/STAT3) transduction pathway. IL-6 acts as a major factor to regulate innate immune response by stimulating the production of MDSCs, neutrophils, macrophages, and C-reactive protein,16–18 as well as by controlling antitumor T-lymphocyte responses.12,19–21 Accordingly, blocking IL-6 signaling may provide a novel therapeutic target and resensitize cancer cells to immunotherapies. For instance, a phase II trial evaluating the clinical benefit of breast and pancreatic cancer patients in the siltuximab (an anti-IL-6 agent) treatment has been completed (NCT00841191).
In this review, we concluded the intracellular IL-6 signaling pathways and described current understandings of IL-6 in tumor immunosuppression. Recent advances in targeted IL-6 therapies in cancers were also summarized.
Structure and intracellular signaling pathways of IL-6
IL-6, which can be secreted by monocytes, T-cells, fibroblasts, and endothelial cells, is a 20-kDa glycoprotein consisting of 184 amino acids which form a typical four-helix structure linked by loops and an additional mini-helix.22,23 The gene encoding IL-6 is located on p14–p21 of chromosome 7. 24 Nuclear factor (NF)-κB upregulates the transcription of IL-6 by binding to its promoter region. 25 Accordingly, the expression of IL-6 can be stimulated by lipopolysaccharide (LPS), tumor necrosis factor-alpha (TNF-α), IL-1α, and all those factors which can activate NF-κB.26,27 In physiological processes, IL-6 mainly induces hematopoiesis, antibody production, and acute-phase reactions via stimulating the production of acute-phase proteins and activating B lymphocytes and T lymphocytes.22,28 Additionally, IL-6 is also involved in regenerative processes, such as neoangiogenesis, in the regulation of metabolism, bone homeostasis, and neural function.29,30
IL-6 receptors embedding on cellular membrane (IL-6R) and soluble IL-6R (sIL-6R) are two existences of receptors for IL-6. IL-6R (also named CD126) and glycoprotein (gp)130 (also named CD130) constitute a cell-surface type I cytokine receptor complex which subsequently activates downstream intracellular signals relevant to proliferation and apoptosis. 24 sIL-6R is derived from transmembrane IL-6R by proteolysis which is mediated via a disintegrin and metalloproteinases 10 (ADAM10) and ADAM17 as well as via mRNA alternative splicing. 31 sIL-6R is mostly detected in peripheral circulation and interacts with IL-6 to activate cells which only express gp130 but not IL-6R.32,33 IL-6/IL-6R is acknowledged as a classic signaling mediating the anti-inflammatory reaction, whereas IL-6/sIL-6R is defined as a trans-signaling endowing IL-6 with proinflammatory characteristics.34,35 On the grounds that the soluble form of gp130 (sgp130) has been demonstrated to be a native inhibitor of trans-signaling, artificially synthesized sgp130-dimers linked by the Fc-part of a human IgG antibody was introduced to clinical trials presently (FE999301).36–38
JAK tyrosine kinase family is found to act as a constitutive kinase domain of gp130 which can be phosphorylated responding to IL-6. 39 Subsequently, activated JAK induces phosphorylated and dimerized STAT3 to translocate to nucleus and bind with DNA elements, resulting in a change in the transcription of numerous genes.40,41 Phosphorylated STAT3 (pSTAT3) is defined as a crucial regulator basically promoting the transcription of apoptosis-related genes, for instance, BcL-XL, MCL-1, c-myc, Fas, and p53. 42 Accordingly, IL-6/JAK/STAT3 pathway is involved in survival, proliferation, and differentiation of normal or tumor cells.24,43 Suppressor of cytokine signaling (SOCS) feedback inhibitors, protein inhibitor of activated Stat (PIAS) proteins, and Src homology region 2 domain-containing phosphatase-1/2 (SHP-1/2) are major mediators restraining the IL-6/JAK/STAT3 pathway under physiological conditions. 44 Moreover, Ras-/mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)/AKT pathways are also two remarkable downstream pathways of IL-6 in inflammation and tumorigenesis processes. Activated Ras protein leads to the phosphorylation of MAPK signaling cascade composed of multiple serine/threonine kinases. And then, MAPK transcriptionally regulates genes associated with cell growth stimulation and the synthesis of acute-phase protein and immunoglobulin.45,46 IL-6-activated JAK also can phosphorylate PI3K enzyme which promotes the generation of phosphatidylinositol-3,4,5-trisphosphate (PIP3) and in turn activates protein kinase B (PkB)/AKT pathway to modify cellular survival signal.47,48 It was reported that PI3K/AKT pathway also relied on IL-6-induced RAS activation in myeloma cells. 49 In general, above three pathways are involved in tumorigenic and anti-apoptotic activities of IL-6, and a schematic signaling diagram is shown in Figure 1. Interrupting any part in this network supplies prospective strategies for cancer treatments.
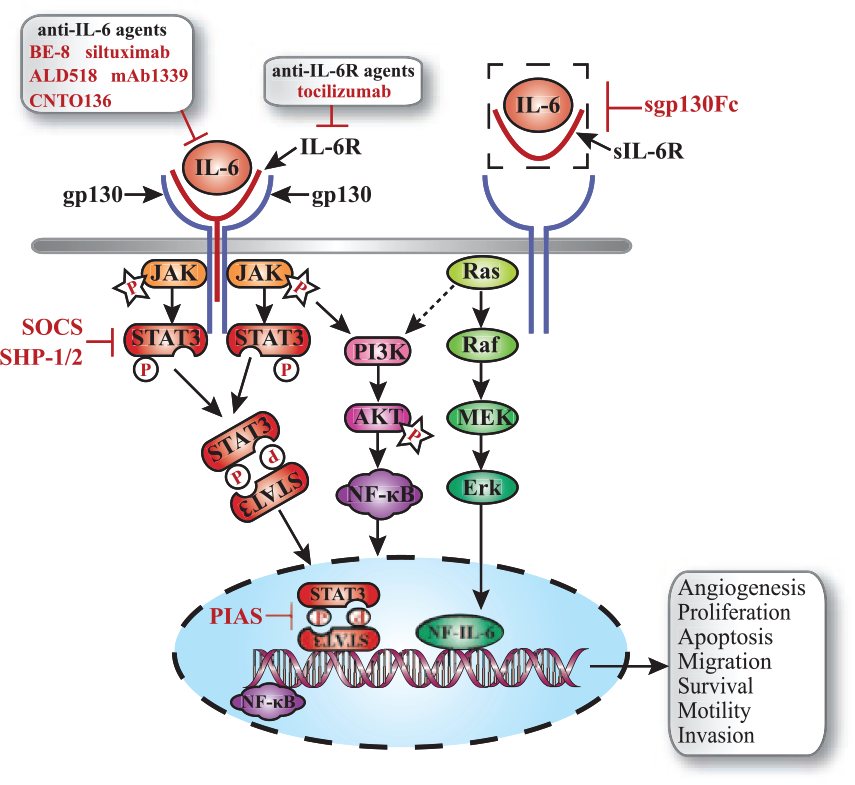
The diagram summarizing the major signaling pathways of IL-6 in cancers. IL-6 interacts with a cell-surface type I cytokine receptor complex, IL-6R/gp130, which subsequently activated intracellular pathways. Responding to IL-6, JAK is phosphorylated and activates STAT3. Phosphorylated STAT3-dimer then transfers to nucleus and binds DNA elements to alter the expression of genes relevant to proliferation, survival, invasion, differentiation, and apoptosis of tumor cells. Another two significant downstream pathways of IL-6 are PI3K/AKT and Ras/MAPK. PI3K/AKT activates NF-κB acting as a transcriptional regulatory factor to promote associated gene expressions. Ras/MAPK regulates genes related to cell growth stimulation and acute-phase protein and immunoglobulin synthesis. Meanwhile, RAS was reported to activate PI3K/AKT pathway in myeloma cells. sIL-6R is a soluble form of IL-6 receptor, and sgp130Fc is a monoclonal antibody invented to block this pathway. Other drugs with ability to block IL-6 pathway include anti-IL-6 agents, anti-IL-6R agents, and STAT3 phosphorylation inhibitor.
The double effects of IL-6 on immunoregulation
IL-6 regulates a vital connection between adaptive and innate immunity mainly by mediating the T cell- and B cell-mediated immune responses. It was found that IL-6 indirectly induced the differentiation of activated B cells into antibody-producing plasma cells, which then synthesize immunoglobulin subclasses, through acting on CD4+ T-cells to upregulate IL-21 production in vitro and in vivo. 50 Consequently, aberrant expression of IL-6 can lead to hypergammaglobulinemia and autoantibody production which contribute to the development of autoimmune and chronic inflammatory diseases. 51 IL-6-defective mice model showed a decrease in B-cell-related immune responses, particularly in the response associated with a T-cell-dependent antigen. 52 IL-6 cooperates with transforming growth factor-β (TGF-β) to stimulate the differentiation of naive T-cell into T helper 17 (Th17) cell which triggers the inflammation reaction and leads to various autoimmune diseases.53–55 There is a positive feedback that IL-6 stimulates Th17 to secrete IL17 which activates NF-κB and in turn to increase the gene expression of IL-6. 56 Whereas IL-6 downregulates the fraction of regulatory T (Treg) cell which restrains the proliferation and related antibody secretion of effector T cell.57,58 Upregulation of the Th17/Treg proportion is involved in destructing immunological tolerance followed by the development of autoimmunity. Thus, IL-6 maintains the dynamic balance between Th17 and Treg. 51 In this aspect, IL-6 can be regarded as a proinflammatory cytokine which is essential for immune homeostasis. 58
In another aspect, IL-6/STAT3 cascade inhibits the expressions of major histocompatibility complex (MHC) class II and CD86 molecules on the surfaces of dendritic cells (DCs) in vivo, resulting in the retardation of cancer-related antigen presentation.59,60 The dysfunction of DCs also attenuates CD4+ T-cell-mediated antitumor immunity responses, and inhibition of IL-6 slows down tumor growth by restoring T-cell activity in tumor-bearing mice model.61,62 Moreover, IL-6 is involved in the differentiation and expansion of MDSCs, which can inhibit T-cell via multiple molecular mechanisms. 63 Overall, IL-6 may play a double role in immunoregulation under pathological conditions.
Interaction between IL-6 and immune resistance in tumor microenvironment
IL-6 and MDSCs
MDSCs, which were described as a heterogeneous population of immature myeloid cells at different stages of differentiation, 64 negatively affected immune responses during inflammation, infection, and cancer via inhibiting T-cell activity and upregulating immunosuppressive cells and cytokines.65–67 Circulating MDSCs were found to be chemoattracted to the tumor site by tumor-derived factors, such as IL-6, chemokine (C-X-C motif) ligand 8 (CXCL8), interferon-γ (IFN-γ), TGF-β, and IL-4. Part of MDSCs could differentiate into tumor-associated macrophages (TAMs) in tumor environment.64,68,69 In addition to the role of immunosuppression, MDSCs have also been identified as a promoter in tumor angiogenesis, invasion, and metastasis. Diaz-Montero et al. 70 investigated that circulating levels of MDSCs were significantly higher in 106 cancer patients with newly diagnosed solid tumors of all stages than in normal volunteers; moreover, among stage IV patients, both percentages and absolute numbers of MDSCs were associated with metastatic tumor burden. Decreased expansion of MDSCs could retard tumor progression by enhancing inflammation process. 71
IL-6 was referred to be a pivotal downstream factor of IL-1β-induced stimulation of MDSCs. Tumor-derived IL-6 compensated the accumulation of MDSCs and was relevant to the primary and metastatic tumor growth in IL-1β-deficient mice. 71 MDSCs also could directly respond to IL-6 via IL-6R which was steadily expressed on their surfaces. 71 Studies revealed that IL-6 produced by tumors was involved in defective myeloid cell maturation and impairment of myelopoiesis which resulted in accumulation and expansion of MDSCs in tumor-bearing hosts mainly via activating JAK2/STAT3 signaling pathway.72,73 In line with this notion, Xu et al. 74 recently showed that drug-resistant hepatocellular cancer cells–derived IL-6 was the major candidate in mediating the expansion and activity of MDSCs using an anti-IL-6 neutralizing antibody that caused a decreased population and immunosuppressive role of MDSCs in vitro culture model and in vivo mouse model. In the 4-NQO-stimulated esophageal squamous cell carcinoma (ESCC) mouse model, IL-6 not only recruited CD11b+CD14+HLA-DR− myeloid cells, a novel subset of MDSCs, but also enhanced the functional activities of MDSCs. Increased production of reactive oxygen species (ROS) and arginase 1 (ARG1), which were involved in MDSCs-induced T-cell inhibition, were detected in IL-6-treated CD14+ cells. 75 In the treated mice, the level of MDSCs was positively associated with the increased possibility of esophageal invasive carcinoma formation, suggesting that IL-6-induced MDSCs might facilitate growth and progression of ESCC in vivo. 75 Moreover, higher levels of IL-6 and CD11b+CD14+HLA-DR− myeloid cells were detected by flow cytometry in the peripheral blood of patients diagnosed with ESCC compared with healthy individuals. 75 In breast cancer, IL-6 activated the STAT3/NF-κB/IDO pathway in MDSCs which was conducive to generate an immunosuppressive tumor microenvironment and lymph node metastasis.76,77 Taking above mechanisms and interactions into consideration, targeting MDSCs and IL-6 might be promising strategies for intervening immunization-mediated tumorigenesis and tumor progression.
IL-6 and TANs
Neutrophils were always recognized as a proinflammatory factor mediating injury restoration and preventing tumor progression via cytotoxic effects as well as vascular endothelium damage. 78 In recent studies, TANs were proposed to promote tumorigenesis and angiogenesis and be associated with poor clinical outcome and heavy tumor burden.79–82 IL-6 and granulocyte-colony stimulating factor (G-CSF) endowed neutrophils with cancer-promoting functions before they infiltrated into the tumor nests not only by modulating related gene expressions but also by reducing neutrophil degranulation. 83 The augmented STAT3 activation and inefficient PI3K and p38 MAPK pathways resulted in reduction of Rab27a in neutrophils, which downregulated the spontaneous release of azurophilic granules from neutrophils. 83 Consequently, costimulation with G-CSF and IL-6 enhanced STAT3 activation and inhibited activation of PI3K and p38 MAPK pathways to increase the expressions of Mmp9 and Bv8 genes and suppress the release of MPO, NE, TRAIL, and defensins from neutrophils, thus attenuated neutrophil degranulation, conducing neutrophils to promote tumor angiogenesis and tumor growth. 83 It was reported that IL-6 secreted by gastric cancer–derived mesenchymal stem cell (GC-MSC) maintained the long-term functional activation of neutrophils through STAT3–extracellular signal–regulated protein kinases 1 and 2 (ERK1/2) pathway, which in turn stimulating the metastatic and angiogenic potentials of gastric cancer and promoting the conversion from normal MSC to cancer-associated fibroblast (CAF) in tumor stroma. 84 Overall, it is reasonable to conclude that IL-6-induced resistance to immune killing occurs mainly by attracting the TANs to tumor milieu and by attenuating and reversing the proinflammatory effects of neutrophils in tumor microenvironment.
IL-6 and epithelial–mesenchymal transition
Epithelial–mesenchymal transition (EMT) is an essential program in multiple developmental processes. In addition to promoting the transition from epithelium to mesenchymal during wound healing and tumor metastasis, EMT was also demonstrated to induce the migration of embryonic epithelial cell during early embryogenesis.85,86 EMT-activated metastatic cascades which contributed to the loss of cell–cell junctions and cell polarity could endow tumor cell with invasive characteristics. 87 EMT is not only sufficient to support primary tumor growth, but may also facilitate the formation and establishment of secondary tumor.
The positive correlation between IL-6 and EMT
The involvements of IL-6 in starting the EMT program and promoting metastatic potential were first shown in estrogen receptor-alpha (ER-α)-positive human breast cancer in 2009. Stable-expressing IL-6 breast cancer cell line (MCF-7IL-6) led to an EMT phenotype characterized by repressed E-cadherin expression and induction of Vimentin, N-cadherin, Snail, and Twist in both RNA and protein levels. 88 Functional assay in vitro certified that IL-6 enhanced the invasiveness and promoted the process of EMT. 88 Similarly, IL-6 overexpressed tumor xenograft also showed EMT changes combined with increased proliferation, advanced tumor stage, and poor histological differentiation in breast cancer.88,89 Remarkably, there was a positive feedback loop in IL-6/STAT3/Twist pathway. IL-6 signaling pSTAT3 to subsequently transactivate Twist which inhibited the transcription of E-cadherin and contributed to EMT. Meanwhile, aberrant accumulation of Twist led to ectopic IL-6 expression and STAT3 activation, suggesting an autocrine manner involved in IL-6-mediated EMT in breast cancer cell.88–90
EMT was generally found to facilitate metastasis in head and neck cancer patients. 91 Yadav et al. 92 first proved the significant role of IL-6 in inducing EMT changes and enhancing metastatic potential via JAK/STAT3/Snail intracellular signal in head and neck squamous cell carcinoma (HNSCC). CAL27 cell stably overexpressing IL-6 exhibited obviously reduction of E-cadherin (53%) and induction of vimentin (54%) and snail (51%). 92 In addition, ectopic IL-6 was relevant to scattering effect which occurred when cell–cell junctions disappeared and was one of the traits of EMT progress. 92 They also discovered that STAT3/Snail pathway stimulated IL-6-mediated EMT in CAL27 cell. 92 Moreover, a severe combined immunodeficient (SCID) mouse xenograft model was utilized to show that higher level of IL-6 enhanced pSTAT3 and Snail proteins to promote EMT and contribute to more distant lymph nodes metastases in HNSCC. 92 Furthermore, IL-6 treatment in cervical squamous cell carcinoma (CSCC) was proved to promote tumor growth, alter cell morphology to mesenchymal, and induce EMT by activating STAT3, especially in poorly differentiated human CSCC tissue. 93
Knockdown of the key transductor implicated in IL-6-induced metastatic cascade can provide an optional regimen for tumor therapies. For example, SHP2, known as an intracellular protein tyrosine phosphatase, is indispensable for the IL-6-induced EMT of breast cancer cells. It was proved that knockdown of SHP2 weakened the IL-6-induced reduction of E-cadherin and IL-6-promoted cell migration and invasion. 94 Metformin, a widely used antidiabetic agent, was found to reverse EMT and attenuate IL-6 signaling in tyrosine kinase inhibitor (TKI)-resistant cells in non-small-cell lung cancer (NSCLC). In xenograft mouse model, utilizing metformin-based combination therapy could block tumor growth which is accompanied by decreased IL-6 signaling activation and EMT. 95 Moreover, by inactivating STAT3 signal, wogonin retarded IL-6-induced EMT both in A549 cell lines and in mice model. 96
IL-6 and cancer stem-like cells
Cancer stem-like cells (CSCs) are specific cells with stem-like properties in cancer cell population. In addition to being possible crucial initiators of cancers, CSCs also enhance the proliferation and invasion of tumor cells and exhibit resistance to conventional antitumor therapies.97–102 IL-6 has been referred to have an effect on the biological characteristics of CSCs. Activation of IL-6-STAT3 pathway was involved in the growth and survival of glioma stem cells (GSCs), which indicated poor outcome for glioblastoma patients. 103 IL-6 receptors (IL6-R and gp130) and ligand showed aberrant expression patterns in GSCs, suggesting that IL-6 could regulate GSCs in both autocrine and paracrine ways. 103 Targeting IL-6R or IL-6 with antibody or shRNA interfered the maintenance and vitality of GSCs in vitro and in vivo. 103 Krishnamurthy et al. 104 have put forward that tumor-associated endothelial cells promoted the survival and self-renewal of CSCs localizing in perivascular niches in patients with HNSCC. In recent research, they found that IL-6 was a crucial mediator in this crosstalk. Endothelial cell–secreted IL-6 enhanced the survival, self-renewal, and invasive potential of HNSCC CSCs via activating STAT3 in a time-dependent manner in vitro. 105 Moreover, in xenograft mouse model, IL-6 also exhibited a vital role in the tumorigenesis of CSCs, and suppression of IL-6 led to a pronounced decrease in the proportion of CSCs. 105 It has been suggested that the IL-6/JAK/STAT3 pathway was preferentially activated and involved in the growth and survival of stem cell–like breast cancer cells in vitro.106,107
Interestingly, EMT was reported to be associated with the generation of CSCs in immortalized human mammary epithelial cells, breast cancer cells, and hepatoma cells.108–111 Xie et al. 112 proved that CD44+ cells with stem-like characteristics stimulated by IL-6/STAT3 also underwent the process of EMT in the epithelial-like T47D breast cancer cells. It indicated that IL-6 might stimulate the generation and maintenance of functional BrCSCs through EMT process, but the explicit mechanisms are still to be determined.
In a recent study, CSCs were identified as critical targets for MDSCs to establish metastasis in the context of breast cancer. MDSCs-derived IL-6 and NO cooperatively activated STAT3 and NOTCH, which supported continuously induction of breast CSCs. 113 The cancer stemness stimulated by MDSCs was conducive to facilitate tumorigenesis, metastasis, and immune escaping which led to poor prognosis in patients with breast cancer. 113 It is tempting to speculate that a complex reciprocal chemokinetic network may exist in tumor microenvironment. CSCs, as a population of the precursors, endow tumor cells with malignant potentials. IL-6 secreted by endothelial cells in tumor-derived neovascular or MDSCs interacts with IL-6R expressed on CSCs to contribute to the generation of CSCs pool, as well as enhance CSCs properties to stimulate invasion and immunosuppression of tumor cells. Targeted one of the elements in this loop may provide promising therapeutic strategies for tumor in the future.
Targeted-IL-6 immunotherapies
Anti-IL6 antibodies
BE-8, a murine anti-IL-6 monoclonal antibody, was investigated in earlier clinical trials. But unsatisfying results were obtained owing to human anti-murine responses and the shorter half-life of BE-8 (3–4 days). 12 After that, a human-murine anti-IL-6 monoclonal antibody, Siltuximab (CNTO 328), with a long half-time (about 2 weeks) and a human IgG1 region to avoid serious immunogenicity, was applied in preclinical and clinical trials ranging from Castleman’s disease to prostate cancer.114–119 In recent 5 years, siltuximab was involved in 13 oncological clinical trials listed as active at Clinical Trials Registry (www.clinicaltrials.gov). The safety, effectiveness, and tolerance of siltuximab were assessed in eight phase I and II studies in patients with newly diagnosed or relapsed or refractory multiple myeloma (MM). Among them, a phase II study, containing 106 patients who were newly diagnosed with MM and randomly received nine cycles of bortezomib–melphalan–prednisone (VMP) alone or VMP plus siltuximab (11 mg/kg every 3 weeks) followed by siltuximab maintenance, suggested that the combination of siltuximab with VMP regimen did not significantly improve the complete response (CR) rate (S + VMP: 27%, VMP: 22%) or overall response rate (S + VMP: 88%, VMP: 80%). 120 The addition of siltuximab to bortezomib in patients with relapsed or refractory MM showed no improvement in progression-free survival (PFS) or overall survival (OS) compared with placebo + bortezomib, despite a numerical increase in response rate in a certain extent. 121 Another three open label phase I/II studies enrolling patients diagnosed with advanced hormone-refractory prostate cancer were conducted to evaluate the safety and efficacy profile of siltuximab as monotherapy or combination therapy. It revealed that siltuximab had favorable tolerance and resulted in a 3.7% prostate-specific antigen (PSA) response rate (RR) and 21% RECIST stable disease rate. 122
CNTO136, ALD518, and mAb 1339 are also characterized as targeted biological anti-IL-6 agents. At the beginning, CNTO 136 and ALD 518 were introduced to clinical trials in patients with autoimmune disease, such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA). 123 But only ALD518 showed well-tolerated and alleviated NSCLC-associated anemia and cachexia in both preclinical and clinical trials. 124 Another promising anti-IL6 agent, mAb 1339, either been used alone or in combination with other agents, exhibited in vitro and in vivo efficacy against MM.125,126
Anti-IL-6R antibodies
Tocilizumab, a humanized anti-IL-6R monoclonal antibody, blocks both sIL-6R and membrane-bound IL-6R, thus inhibits both classic signaling and trans-signaling. In the last decade, tocilizumab has already been approved for the treatment of Castleman’s disease, RA, systemic-onset, and polyarticular-course juvenile idiopathic arthritis.127–129 Besides potential roles of tocilizumab in IL-6-driven autoimmune and chronic inflammatory diseases, promising effects on malignant diseases such as ovarian cancer, oral cancer, glioma, MM, and mesothelioma were also found. 130
In 2016, there were six clinical trials listed as not yet recruiting, recruiting, or completed at www.clinicaltrials.gov about the utilization of tocilizumab in cancer patients. A completed phase I trial evaluated the feasibility, safety, and immunoenhancement of the combination of chemotherapeutics (Carbo/Caelyx or doxorubicin), blockade of IL-6R (tocilizumab), and immune enhancer interferon-α (Peg-Intron) in patients with recurrent epithelial ovarian cancer (EOC). It showed that the addition of tocilizumab in carboplatin/pegylated liposomal doxorubicin (PLD) was feasible and safe in EOC patients. 131 And highest dosage of tocilizumab (8 mg/kg) effectively blocked IL-6R with increased levels of serum IL-6 (p = 0.02) and soluble IL-6R (p = 0.008). 131 Interestingly, the EOC patients with a significant increase in sIL-6R showed a longer median OS time (p = 0.03). 131 The obstruction between IL-6R and IL-6 contributed to decreased pSTAT3 in immune cells, increased IL-12 and IL-1β produced by myeloid cells, and larger quantities of immunological cytokines (IFN-γ and TNF-α) secreted by activated T-cell.
General anti-IL-6 agents
Some chemical agents, such as corticosteroids, NSAIDs, and statins, exert inhibitory effect on IL-6 by controlling its expression through blocking NF-κB. Those agents were widely applied as adjuvant or synergistic drug in amounts of malignancies. Dexamethasone was investigated to suppress the growth of MM partly via the inhibition of IL-6 production and had the potential to be used in hormone-refractory prostate cancer and advanced renal cell carcinoma therapy.132–134 Even so, the serious side effects of corticosteroids lead to its limitations in therapeutic use. NSAIDs, particularly aspirin, were mostly introduced to prevent or treat various kinds of cancers. Usage of NSAIDs on a regular basis decreased the risk of breast cancer via modifying the IL-6 genotype and of ovarian cancer via promoting apoptosis and restraining angiogenesis.135,136 NSAIDs also enhanced antitumor immunity by inhibiting IL-6 in human hepatocellular and oral squamous cell carcinoma.137,138 Simvastatin, as a member of statins, was revealed to inhibit tumor growth and metastasis by suppressing the phosphorylation of JAK2/STAT3 activated by IL-6 in renal cell carcinoma. 139
Conclusion
To date, IL-6 and its related signal pathways have been well defined. Elevated level of IL-6 in various malignancies is involved in tumor growth and metastasis, thus inducing to a higher tumor burden which includes advanced tumor stage, shortened survival, and earlier recurrence. Notably, IL-6 is associated with chemo-resistance which is one of the most difficult impediments to cure cancer via inhibiting the immune responses and promoting the immune escaping of tumor cells. Based on its roles in immune-modulating, IL-6 can alter the tumor microenvironment toward immunosuppression by attracting and activating associated factors, such as MDSCs, TANs, Tregs, and CSCs. And the schematic diagram showed an immunosuppressive network centered on IL-6 in tumor microenvironment in Figure 2. Therefore, targeted inhibitor of IL-6 has become a crucial class of antitumor drugs in recent years. More and more preclinical and clinical trials have been conducted to evaluate the availability of anti-IL-6 agents in cancers. It may offer molecular targeted therapeutic strategies to sensitize tumor cells to chemotherapy and eventually improve the prognosis of patients with malignancies. It is also remarkable that blockage of IL-6 transduction pathways may be potentially useful in clinical therapy. Nevertheless, the dosage and side effects of anti-IL-6 agents, and whether they can be used alone or in combination with conventional chemotherapy, must be investigated in the future.

Immune resistance caused by IL-6 within tumor microenvironment. MDSCs and TANs can be chemoattracted to tumor nests by IL-6 from peripheral circulation and contribute to immunosuppression. Attracted MDSCs differentiate into TAMs in tumor nests. IL-6 secreted by tumor cells and MDSCs binds to IL-6R expressed on the surface of above two cells to present both autocrine and paracrine manners. Endothelial cells of tumor-derived neovascularization also express IL-6, which interacted with the IL-6R on CSCs to result in the generation of CSC pool and maintain the CSC properties.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This review was supported by National Natural Science Foundation of China (Grant No. 81572608) and the National High Technology Research and Development Program of China (Grant No. 2015AA020301).