
Abstract
JNKs (c-Jun N-terminal kinases) belong to mitogen-activated protein kinases’ family and become activated by several growth factors, stress, radiation, and other extracellular signals. In turn, JNK activation results in phosphorylation of downstream molecules involved in many normal cellular processes. Nevertheless, recent data have linked JNK signaling with several pathological conditions, including neurodegenerative diseases, inflammation, and cancer. The role of JNK in cancer remains controversial. Initially, JNK was thought to play a rather oncosuppressive role by mediating apoptosis in response to stress stimuli, inflammatory, or oncogenic signals. However, a number of studies have implicated JNK in malignant transformation and tumor growth. The contradictory functions of JNK in cancer may be due to the diversity of JNK upstream and downstream signaling and are under intensive investigation. This review summarizes current literature focusing on the significance of JNK pathway in cancer development and progression, particularly addressing its role in oral cancer. Understanding the complexity of JNK signaling has the potential to elucidate important molecular aspects of oral cancer, possibly leading to development of novel and individualized therapeutic strategies.
Keywords
Introduction
c-Jun N-terminal kinases (JNKs) belong to the family of mitogen-activated protein kinases (MAPKs) and are involved in a wide spectrum of cellular processes, including cell proliferation, differentiation, migration, inflammation, and apoptosis.1–3 JNK was first recognized as a stress-activated protein kinase (SAPK) in mice liver under the term cycloheximide, 4 but then named “JNK” to emphasize its role in phosphorylation and activation of c-jun transcription factor. In human genome, JNK kinase family includes three members encoded by three genetic loci, known as JNK1 (Mapk8), JNK2 (Mapk9), and JNK3 (Mapk10). 5 Whereas JNK1 and JNK2 are ubiquitously expressed in most tissues, JNK3 expression is predominantly found in brain, as well as in heart and testes though to a lesser extent. 6 Similar to other MAPKs, JNKs are strongly activated in response to cytokines, ultraviolet (UV) radiation, stress, and reagents that cause DNA damage and less frequently after stimulation of G-protein-coupled receptors (GPCRs), serum, and growth factors.7–9
JNK pathway is activated by two upstream mitogen-activated protein kinase kinases (MAP2Ks) (MKK4 and MKK7) that directly phosphorylate JNKs on threonine (Thr183) and tyrosine (Tyr185) residues. 3 In turn, MKK4 and MKK7 are activated by upstream pathways through various mitogen-activated protein kinase kinase kinases (MAP3Ks) including MEKK1-4, MLK2 and -3, Tpl-2, DLK, TAO1 and 2, TAK1, and apoptosis signal-regulating kinase (ASK)1/2. 10 Each MAP3K could be specific to different stimuli. For example, TAK1 is important for activation of JNK in response to inflammatory cytokines (interleukin-1 (IL-1), tumor necrosis factor-alpha (TNF-α), transforming growth factor (TGF)-β, and lymphotoxin-b) 11 and for activation of toll receptors (TLR-3, -4, and - 9).12,13 MEKK3 is important for activation in response to TLR-8, while MAP3K isomers (Tpl-2 and MLK-3) have been reported to take part in TNF-dependent activation of JNK in fetal fibroblasts14,15 (Figure 1).
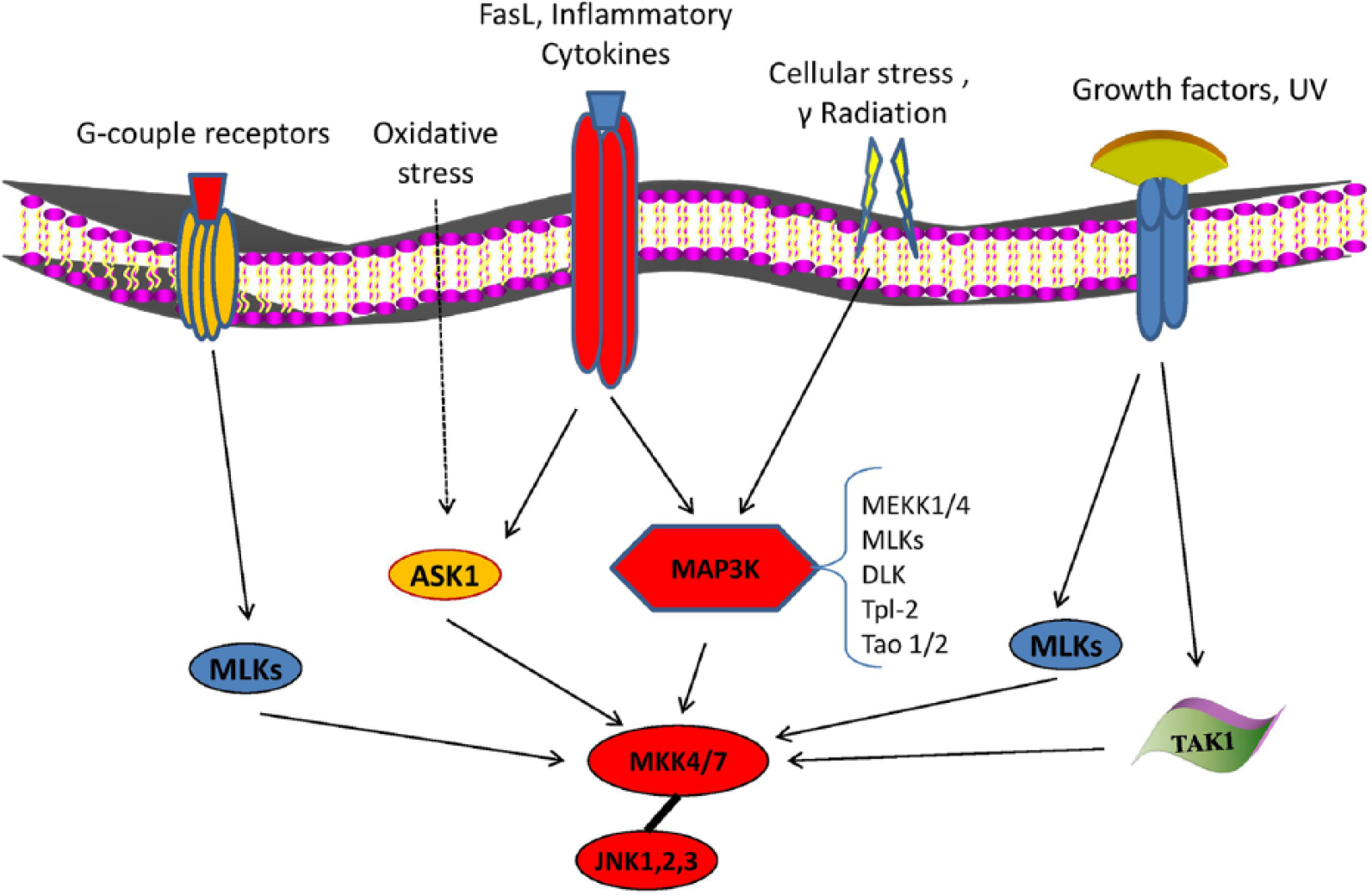
Activating mechanisms of JNK signaling. JNKs are strongly activated in response to cytokines, UV radiation, stress, and reagents that cause DNA damage (γ-radiation) and less frequently, after stimulation of G-protein-coupled receptors and growth factors. JNK pathway is activated by two upstream MAP2Ks (MKK4 and MKK7) which directly phosphorylate JNKs on threonine (Thr183) and tyrosine (Tyr185) residues. In turn, MKK4 and MKK7 are activated in upstream pathways by various MAP3Ks including MEKK1-4, MLK2 and -3, Tpl-2, DLK, TAO1 and 2, TAK1, and ASK1/2, which could be specific to different stimuli.
A large number of nuclear proteins, mainly transcription factors and nuclear hormone receptors which regulate a plethora of cellular activities, such as proliferation, differentiation, survival, and apoptosis have been identified as JNK substrate proteins (Figure 2). Jun proteins, JunB, JunD, and particularly c-jun are the most important nuclear substrates, which, once activated, form the transcription factor activator protein-1 (AP-1) after dimerization with Fos proteins (c-Fos, FosB, Fra-1, and Fra-2). 6 Other JNK downstream signaling molecules include activating transcription factor 2 (ATF-2), c-Myc, p53, Elk1, nuclear factor of activated T cell (NFAT), signal transducers and activators of transcription (STAT)1 and 3, and the family protein Pax, 6 as well as mitochondrial apoptosis regulators of Bcl-2 family (Bcl-2, Bcl-xl, Bad, Bim, and Bax). 3 Complexity of JNK signaling plays significant role in normal cellular functions including controlled degradation of proteins, 16 cell and tissue morphogenesis, and immune response. 17 Moreover, several studies have highlighted the role of JNK activity in the pathogenesis of several diseases, such as diabetes, 18 lung fibrosis, inflammatory and neurodegenerative disorders,19,20 as well as cancer.5,21

Downstream target molecules of JNK signaling pathway. Jun proteins (c-Jun, JunB, and JunD) and particularly c-jun are the most important JNK nuclear substrates, which, once activated, form transcription factor activator protein-1 (AP-1) after dimerization with Fos proteins (c-Fos, FosB, Fra-1, and Fra-2). Other JNK downstream signaling molecules include activating transcription factor 2 (ATF-2), SMAD4, p53, Elk1, NFAT, and STAT3. These indicative substrates regulate a plethora of cellular activities, such as proliferation, differentiation, survival, and apoptosis, as well as immune response.
JNK role in cancer
The role of JNK in cancer remains controversial. Initially, JNKs were thought to play a rather oncosuppressive role by promoting apoptotic cell death in response to stress stimuli, inflammatory, or oncogenic signals. 22 However, more recent data suggested that JNKs and especially JNK1 contribute to malignant transformation and tumor growth. 5 Indeed, several studies demonstrated that upregulation of JNK enhances tumor growth or induces drug resistance in various cancer cell lines, including hepatocellular carcinoma (HCC), cholangiocarcinoma, anaplastic large-cell lymphoma, and breast cancer.23–28
Additionally, it is widely known that malignant transformation induced by Ras oncogene is regulated by c-Jun, a direct substrate of JNK. 29 In agreement with the oncogenic role of JNK, it was reported that JNK1, but not JNK2, is overexpressed in primary HCCs compared to non-cancerous tissues,26,30 while JNK1 was found to regulate human HCC cell proliferation by affecting p21 and c-Myc expression. 26 Moreover, Chang et al. 30 suggested that high JNK1 expression levels correlated with poorer prognosis in patients with HCC, and Takahashi et al. 31 demonstrated that JNK1 promoted tumorigenic activity of tobacco smoke in the lungs.
Furthermore, recent studies have highlighted the oncogenic potential of JNK2 in several human cancer cells, including glioblastoma, lung, and prostate carcinoma cells,32–34 as well as in an animal squamous cell carcinoma model in which JNK2, but not JNK1, interacted with Ras and induced malignant transformation of primary human epidermal cells. 35 Furthermore, Barbarulo et al. 36 showed that JNK2 was essential for survival of myeloma cells and suppressed JNK1-mediated apoptosis by increasing expression of poly(ADP-ribose) polymerase family member 14 (PARP 14; regulator of B-cell survival) in multiple myeloma (MM). In addition, Cellurale et al. 37 concluded that both JNK1 and 2 promoted Ras-induced lung carcinogenesis in mice models characterized by mutational activation of the endogenous KRas gene.
However, JNK is a known key regulator of stress-induced apoptosis and autophagy, as well as TNF-α or UV-induced apoptosis.22,38 Bcl-2 and Bcl-xl inhibition or Bax activation are some of the apoptotic mechanisms enhanced by JNK upon its activation.39–41 In line with its positive contribution to apoptosis, several lines of evidence suggested a tumor-suppressive role of JNK signaling in cancer.42,43 For example, She et al. 44 reported that JNK1-deficient mice are more susceptible to tetradecanoylphorbol-13-acetate (TPA)-induced skin tumor development than wild-type mice and proposed JNK1 as a negative regulator of tumorigenesis. In addition, experimental JNK1-null mice were highly susceptible to both melanoma or lymphoma tumorigenesis compared with wild-type models as a result of functional defects of CD8+ T cells. 45 In accordance to the findings in animal models, several studies in human cancer cells proposed an anticancer role of JNK activation. Indeed, Chauhan et al. 46 demonstrated that transfection of MM cells by dominant-negative JNK mutant or treatment with JNK chemical inhibitor abolished stress-induced release of Smac and apoptosis. Furthermore, JNK activation induced paclitaxel (tubule poison) phosphorylation of Bcl-2 resulting in increased breast cancer cell apoptosis. 47 Similarly, impaired JNK signaling appeared to enhance tumor development in prostate, breast, or pancreatic cancer cells,48–50 while inhibition of MKK7-JNK pathway by TOR signaling pathway regulator-like (TIPRL) protein prevented TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in HCC cells. 51 In addition, Oleinik et al. 52 reported that JNK2 phosphorylated the tumor-suppressor p53 on Ser6 residue, while silencing of both JNK1 and JNK2 abrogated apoptosis of non-small-cell lung carcinoma cells (Table 1).
Representative examples of specific types of cancer in which JNK exerts either oncogenic or oncosuppressive roles, including related upregulated (↑) or downregulated (↓) molecules.

TNF: tumor necrosis factor; TGF: transforming growth factor; mTOR: mechanistic target of rapamycin; IL: interleukin; STAT: signal transducers and activators of transcription.
Role of JNK in oral cancer
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer in developed countries with continuing rise of new cases and a low 5-year survival rate. 53 Recent studies have focused on JNK signaling in oral cancer, and emerging data give new insight into the role of JNK pathway in this type of cancer.
Oncosuppressive role of JNK in oral cancer
A substantial body of evidence has implicated JNKs as tumor suppressors in oral cancer, emphasizing their positive role in apoptosis. Accordingly, several studies have proposed that JNK may act alone or synergistically with other MAPKs in regulating oncogenic or oncosuppressive pathways. For example, Noutomi et al. 54 reported that JNK activation is involved in TRAIL-induced apoptotic cell death in oral squamous cell carcinoma (OSCC) cells. Next, Kim et al. 55 proposed that activation of JNK, p38, and extracellular signal–regulated protein kinase (ERK) MAPKs, as well as protein kinase C (PKC), mediates N-(4-hydroxy-phenyl) retinamide-induced apoptosis triggered by reactive oxygen species (ROS) in OSCC. Other relevant studies also examined polyphyllin G-antitumor effect suggesting that upregulation of ERK, p38, and JNK pathways mediated the activation of Akt-dependent apoptotic signaling in OSCC and nasopharyngeal cancer cells.56,57 Likewise, Kurihara et al. 58 described that integrin-α5 knockdown downregulated JNK activation and reduced apoptosis, while p38 suppression increased cell death in OSCC.
More recently, several studies examined a possible crosstalk between JNK and major signaling molecules in oral cancer, including signal transducers and activators of transcription (STATs) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). Specifically, interferon (IFN)-γ-treatment of OSCC cells activated Janus kinase (JAK)/STAT1, p38, JNK, and NF-κB and induced apoptosis through an endoplasmic reticulum (ER) stress response modification. 59 Another in vitro study suggested an oncosuppressive role of JNK1/2 signaling in OSCC, probably mediated through negative crosstalk with the oncogenic STAT3 pathway. 60 In addition, Lee et al.61–63 conducted a series of studies concerning the tumor-suppressive role of bioactive phytochemical components in HN4 and HN12 oral cancer lines. They first examined the apoptotic effect of two compounds extracted from Caesalpinia sappan (sappanchalcone and isoliquiritigenin 2′-methyl ether (ILME)) and found that sappanchalcone downregulated cell proliferation and induced apoptosis through mechanisms mediated by p53 and dependent upon p38, ERK, JNK, and NF-κB signaling; 61 furthermore, ILME induced expression of p21 and p53 and activated NF-κB, ERK, and JNK. 62 Later on, the same investigators showed that mollugin (extracted from Rubia cordifolia) induced apoptosis and increased expression of p38, ERK, and JNK, while suppressed activation of NF-κB and NF-κB-dependent antiapoptotic products (Bcl-2 and Bcl-xl). 63 Correlation between NF-κB and JNK signaling in oral cancer was also reported by Cacalano et al. 64 who demonstrated that the gene and protein expression levels of insulin-like growth factor binding protein 6 (IGFBP6) were elevated by the synergistic effects of JNK overexpression and NF-κB inhibition accompanied by a significant enhancement of apoptosis in a mouse model of oral tumor cells. In contrast, Chen et al. 65 suggested a synergistic interaction, in which both NF-κB and JNK were essential for apoptosis induced by aminolevulinic acid in human oral cancer (Ca9-22) cells.
Furthermore, previous investigations have focused on the individual role of JNK in oral cancer cell apoptosis, in combination with various chemotherapeutic reagents. For example, Li et al. 66 examined the antitumor ability of mechanistic target of rapamycin (mTOR) inhibitor (AZD8055) and reported that AZD8055 significantly induced JNK activation and increased cell death through autophagy in HNSCC lines (Hep-2 and SCC-9). Interestingly, JNK chemical inhibition or silencing by RNA interference reversed AZD8055-induced apoptotic effects. 66 Moreover, Masuda et al. 67 suggested that combinatory treatment of two HNSCC cell lines (YCU-N861 and YCU-H891) with 5-fluorouracil (5-FU) and all-trans-retinoic acid (ATRA) resulted in synergistic JNK1 activation and further enhanced apoptosis. Cisplatin and cordycepin were also proposed to induce synergistically apoptosis by activating JNK/caspase-7/PARP cascade in OC3 oral cancer cell line. 68 In addition, green tea polyphenol epigallocatechin-3-gallate (EGCG) is already reported to inhibit cell growth and induce caspase 3-mediated apoptosis in OSC2 cell line. 69 To determine JNK role in this event, Yamamoto et al. 70 produced a p57-deficient OSC2 cell line (p57 gene is often associated with advanced tumors) and showed that in modified cell lines, JNK activation mediated EGCG-induced apoptosis, while exogenous p57 administration inhibited EGCG-induced proapoptotic JNK signaling in OSC2 cells. In addition, silencing of XBP1 protein along with MG132 (protease inhibitor) treatment of OSCC cells further blocked XBP1 survival pathway and induced apoptosis through activation of ASK1-JNK signaling. 71
Interestingly, previous studies attempted to establish synergistic or antagonistic effects between JNK and other MAPKs; however, results remain controversial and inconvenient to determine a secure relation. For example, synergistic role of p38 and JNK in HNSCC apoptosis was reported in an in vitro study as a response to excessive ROS production of cancer cells after acetylshikonin treatment. 72 Moreover, overexpression of ERK, JNK, and p38 was suggested to regulate (6-(N,N-Dimethylamino)-2-(naphthalene-1-yl)-4-quinazolinone; DPQZ)-induced apoptosis in HSC-3 cells. However, within MAPKs, only inhibition of JNK alleviated DPQZ-induced cell death as well as the increase in Bcl-2, caspase-3, caspase-8, and caspase-9 activation. 73 Similarly, treatment of OSCC cells with either PCH4 (n-butylidenephthalide-derivative) 74 or griseofulvin 75 activated JNK but not ERK or p38, induced apoptosis, and resulted in Bcl-2 phosphorylation. 75 Also, JNK pharmacological inhibition diminished Lactoferrin-induced apoptosis 76 and inhibited bortezomib-mediated autophagy 77 in oral cancer cells, whereas ERK1/2 76 or p38 77 inhibitors showed no effect on apoptosis, but rather an increase in cell death. 76 In agreement, gamma-aminobutyric acid (GABA) and Muscimol (GABA A type receptor agonist) enhanced cell proliferation and decreased OSCC cell apoptosis by activating p38 MAPK and downregulating JNK signaling pathway. 78 Ghose et al. 79 also reported that JNK pathway is the most critical for Fas-L activation, induction and apoptosis as inhibition of JNK, but not of ERK1/2, downregulated Fas-L expression and cell death, while Yang et al. 80 showed that activation of JNK combined with downregulation of ERK1/2 enhances the apoptotic effect on oral cancer cells. In contrast, Antrodia camphorata (AC) 81 and fomitoside-K 82 were found to induce phosphorylation of both ERK1/2 and JNK and triggered apoptosis in OSCC cell lines.
In addition, other researchers investigated the role of MAPKs including JNK in oral cancer cell proliferation and migration. Hence, treatment of SCC-4 and SAS cancer cells with either phenethylisothiocyanate (PEITC) 83 or berberine (isoquinoline alkaloid) 84 increased JNK, ERK, and p38 phosphorylation, inhibited epidermal growth factor receptor (EGFR) signaling, 83 reduced expression of both MMP-2 and MMP-9, and prevented migration and invasion of tumor cells.83,84 Furthermore, inhibition of hepatocyte growth factor was associated with downregulation of AKT and ERK downstream kinases as well as with upregulation of p38 and JNK, followed by decreases in tumor growth, invasion, and MMP-2/MMP-9 expression in KB oral cancer cells. 85 In line, Husvik et al. 86 proposed that JNK downregulated EGF-induced ERK1/2 expression and p38-mediated COX-2 transcriptional activity in two oral cancer cell lines. Moreover, Yen et al. 87 suggested that treatment of oral cancer cells with cardiotoxin III reduced MMP-2 and MMP-9 protein expression, increased JNK and p38 activation, and repressed proliferation and migration, while JNK inhibition abolished 1α,25(OH)2D3-mediated antitumor activity in three oral cancer cell lines (Table 2). 88
Synopsis of the available literature data concerning JNK oncogenic or oncosuppressive roles in oral cancer, including related upregulated (↑) or downregulated (↓) molecules and highlighting the major biologic effect.

VEGF: vascular endothelial growth factor; EGFR: epidermal growth factor receptor; IL-8: interleukin-8; MMP: matrix metalloproteinase; FAK: focal adhesion kinase; STAT: signal transducers and activators of transcription; ROS: reactive oxygen species; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells.
Tumorigenic role of JNK in oral cancer
Although JNK was primarily considered to play an antitumor role in oral cancer, overwhelming evidence has unequivocally unraveled an opposite effect, based on which JNK promotes tumorigenesis. Specifically, Kuo et al. 89 proposed that c-Jun activation could serve as a prognostic marker of tumor progression in oral SCC. Furthermore, JNK inhibition was found to downregulate IL-8 signaling as well as vascular endothelial growth factor (VEGF) and EGFR activation in oral cancer cells, resulting in reduction of cell proliferation and capillary tube formation thus suppressing tumor growth. 90 Similarly, combined inhibition of BAG3 and JNK in HSC-3 oral cancer cells contributed to better tumor response in hyperthermia therapy, 91 while knockdown of Twist or JNK inhibition enhanced cisplatin-induced apoptosis in both SCC-4 and Tca8113 oral cancer cells. 92 Chai et al. 93 also reported that combined treatment of SAS cells with JNK inhibitor and acridine-based N-acyl-homoserine lactone (AHL) analog prevented mitosis and resulted in atypical mitotic cell death.
In addition, recent evidence examined the contribution of MAPKs expression, including JNK, in oral cancer cell invasion and metastasis. For example, Chang et al. 94 suggested that combinatory treatment of CAL-27 cells with EGCG and gefitinib downregulated phosphorylated levels of EGFR as well as phosphorylated ERK, JNK, p38, and AKT and repressed the metastatic potential of tumor cells. Sento et al. 95 proposed that OSCC cells secreted exosomes which in turn were uptaken by adjacent recipient OSCC cells, driving to enhanced proliferation, migration, and invasion through ERK- and JNK1/2-dependent pathways. Moreover, treatment of oral cancer cells with glial cell–derived neurotrophic factor (GDNF) increased expression of MMP-9 and MMP-13, enhanced activation levels of ERK, p38, and JNK, as well as AP-1 DNA binding activity; and induced cell migration ability. 96 Also, Chuang et al. 97 indicated that IL-6 enhanced migration of oral cancer cells by upregulating Syk/JNK/AP-1 signaling pathway. Furthermore, microRNA-196 (miR-196) was found to activate phosphorylated JNK and MMP1/9, thus enhancing cell migration and invasion, 98 while focal adhesion kinase (FAK) downregulation or knockdown quenched the metastatic potential of oral cancer cells also reducing expression of MMP2/9, phospho-JNK, and phospho-p38.99,100 Additionally, another study examined the anti-invasive activity of the naturally occurring agent resveratrol, which downregulated both phospho-JNK and phospho-ERK1/2 followed by inhibition of MMP-9 transcription. 101 Moreover, IL-20 was found to promote proliferation and migration in oral cancer cells through STAT3 activation and induction of AKT/JNK/ERK signaling pathway. 102 Similarly, Fos-like antigen 1 (FRA1) was found to enhance cell growth by triggering AKT signaling, as well as cell migration through JNK/c-Jun pathway in SCC of the skin and the head and neck (Table 2). 103
Conclusion
This review underscores the dual role of JNK1/2 in oncogenesis; obviously, these molecules possess both tumor-promoting and/or tumor-suppressor functions in different types of malignant cells, including oral cancer. Several theories have attempted to provide an explanation for the contradictory functions of JNK: it has been proposed that simultaneous induction of growth promoting and apoptotic pathways may occur as a result of compensatory proliferation of neighboring cells promoted by the apoptotic stressed cells 5 or that JNK-induced autophagy could act as tumor suppressor in early oncogenic transformation, while promoting survival in established tumors. 104 Furthermore, it has been speculated that prolonged JNK activation exerts proapoptotic effects, while transient activation rather enhances cell survival. 105 Notwithstanding the gaps in existing knowledge, it is now widely accepted that JNK activity in cancer is tissue specific and cell type-dependent; moreover, JNK signaling may vary according to tumor stage, status, and availability of activated upstream and downstream molecules and a plethora of stress signals. Understanding the complexity of JNK pathway and its crosstalk with other crucial molecules is a major challenge as well as an opportunity to develop more specific and individualized anticancer therapeutic strategies.
Footnotes
Acknowledgements
All authors listed above have contributed significantly to implementation, analysis, interpretation of the data, and general presentation of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.