
Abstract
The incidence of disease relating to nanoparticle exposure has been rising rapidly in recent years, for which there is no effective treatment. Macrophage is suggested to play a crucial role in the development of pulmonary disease. To investigate the changes in macrophage after being stimulated by nanometer silica dust and to explore potential biomarkers and signaling pathways, the gene chip GSE13005 was downloaded from Gene Expression Omnibus database, which contained 21 samples: 3 samples per group and 7 groups in total. Macrophages in the control group were cultured in serum-free medium, while the experimental groups were treated with nanometer silica dust in different sizes and concentrations, respectively. To identify the differentially expressed genes and explore their potential functions, we adopted the gene ontology analysis and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis and also constructed protein–protein interaction network. As a result, 1972 differentially expressed genes were identified from 22,690 microarray data in the gene chip, 1069 genes were upregulated and 903 genes were downregulated. Results of the gene ontology analysis indicated that the differentially expressed genes were widely distributed in intracellular and extracellular regions, regulating macrophage apoptosis, inflammatory response, and cell differentiation. The Kyoto Encyclopedia of Genes and Genomes pathway analysis showed that the majority of differentially expressed genes were enriched in cytokine–cytokine receptor interaction, cancer or phagosome transcriptional misregulation. The top 10 hub genes, S100a9, Nos3, Psmd14, Psmd4, Lck, Atp6v1h, Jun, Foxh1, Pex14, and Fadd were identified from protein–protein interaction network. In addition, Nos3, Psmd14, Atp6v1h, and Jun were clustered into module M2 (rc = 0.74, p < 0.01), which mainly regulates cell carcinogenesis and antivirus process. In conclusion, differentially expressed genes screened from this study may provide new insights into the exploration of mechanisms, biomarkers, and therapeutic targets for diseases relating to nanoparticle exposure.
Introduction
With the wide application of nanomaterial, nanoparticle exposure has been becoming a common phenomenon in daily life. More and more studies have indicated that nanometer silica dust exposure is strongly associated with pulmonary inflammatory response, interstitial fibrosis, and lung cancer.1–3 Similar to other dusts, nanoparticles involved in human body mainly depend on respiratory inhalation; due to the tiny size, the inhaled nanoparticles can hardly be exhaled. As a result, most of the particles accumulated in pulmonary alveoli, what is worse, a small amount of particles can directly go through the air–blood barrier, arriving in peripheral blood via pulmonary circulation and causing multi-organ disorders.4,5 Current studies have found that alveolar macrophage (AM) plays a crucial role in foreign body phagocytosis. Consequently, silica dusts accumulated in alveoli would be identified and swallowed by AM, initiating the antigen-presenting process to immune cells.6,7 Along with silica dust accumulation, it may induce AM apoptosis or autophagy, besides, the ruptured macrophages may release a variety of cytokines and chemokines into pulmonary interstitial space, activating pulmonary inflammatory response.8,9 In addition, the stimulation induced by inflammatory factors further induces lung fibroblast transdifferentiating into myofibroblast. As a result, a large amount of collagen was synthesized and deposited in the extracellular matrix, which could subsequently promote pulmonary fibrosis.10–12 Although studies at the cell level have preliminarily revealed the pathogenic process associated with nanoparticle exposure, the integrated mechanism still needs further exploration.
Advances in molecular biology and genomics make the further exploration possible, high throughput sequencing technology has been widely used in disease diagnosis and prognosis. Recently, some hub genes related to lung cancer have been approved, but nanoparticle-induced gene expression profile changes were rarely discussed. Waters et al. 13 found that the gene expression profile of macrophages was highly conserved among stimulations induced by different particle sizes, but few differentially expressed genes (DEGs) were proposed limited by the old database and bioinformatics methods, and the protein–protein interaction (PPI) network was also deficient. However, the phagocytosis of nanoparticles by macrophage plays an important role in pneumoconiosis and lung cancer development, so it would be of great significance to identify DEGs in nano-silica exposed macrophages. In this study, we integrated the DNA microarray technology and bioinformatics technology comprehensively and subsequently identified the key DEGs and signaling pathways; furthermore, we explored the role of DEGs in pulmonary fibrosis development from three aspects, including cellular components, molecular function, and biological process, which were also used to mine potential pathogenic genes and biomarkers.
Materials and methods
Microarray data
Gene chip GSE13005 was downloaded from the Gene Expression Omnibus (GEO) database, which was based on Agilent GPL6480 platform (Affymetrix mouse genome 430A 2.0, Mouse430A_2), contained 21 samples: 3 samples per group and 7 groups in total. RAW 264.7 mouse macrophages in the control group were cultured in serum-free medium, while the experimental groups were exposed to 10 nm silica at 5 (low), 20 (mid), or 50 (high) µg/mL or 500 nm silica at 250 (low), 500 (mid), or 1000 (high) µg/mL for 2 h. As concluded by Waters et al., 13 the changes in the macrophages gene expression profile were highly conserved among different stimulations; here, we integrated all experimental groups as silica stimulating group and then made a comparison with the control group for the next step data mining.
DEGs screening and function exploration
First, SAS 9.2 was used to preprocess the raw data collected from GEO database, RMAExpress software was used to do background adjustment, quantile normalization, and summarization. After matching to the annotation file which was downloaded from the Affymetrix official website (http://www.affymetrix.com), the combined dataset was outputted, which included microarray data and official gene symbols. In the next step, we hierarchically clustered samples and genes respectively with the average linkage method, signal to noise was selected as the test-statistic assessment indicator, and three conditions were set for DEGs screening: false discovery rate (FDR) <20%, unfolded change >2, and p value <0.01. After logarithmic transformation, the DEGs expression correlationship between the control group and the experimental group was evaluated. Gene ontology (GO) analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment were conducted under the preset conditions (thresholds account ≥2 and ease score ≤0.1) with Database for Annotation, Visualization and Integrated Discovery (DAVID) 6.8 online (https://david.ncifcrf.gov/).
PPI network and module clustering
Search Tool for the Retrieval of Interacting Genes (STRING) database is a biological database and web resource of known and predicted PPI which covers 184 million interaction pathways constructed by 9.6 million proteins from 2031 organisms. In this study, the combined score was set as 0.4 to screen DEGs, and the PPI dataset was clustered with the Molecular Complex Detection (MCODE) plug-in in Cytoscape 3.4.0; the cluster condition was set as follows: haircut mode, node score cutoff value 0.2, and K-core value 2. Moreover, all modules were screened with a cluster coefficient value ≥0.5 if not specified.
Results
1972 DEGs were identified
In total, 21 samples were included in the analysis: 3 samples in the control group and 18 samples in the experimental group. Data from each gene chip were merged into a consolidated database, the Gene-e software was used to identify DEGs, and three conditions were set for DEGs screening: FDR <20%, unfolded change >2 and p value <0.01. A total of 1972 genes were proposed after the analysis, of which, 1069 were upregulated and 903 were downregulated (Figure 1(c)). Moreover, the gene expression correlation analysis was conducted with the top 100 genes; the correlation coefficient was 0.9932 and shown in Figure 1(a) and (b) (p < 0.001).

The screening results of differentially expressed genes (DEGs). (a) The correlation analysis between the experimental group and the control group; the top 100 DEGs were used to construct the correlation analysis model, the correlation coefficient was 0.9932 (p < 0.001). (b) The gene expression profile comparison between the experimental group and the control group, the value of mean, median, upper, and lower quartiles of the experimental group (red) were all higher than the control group (blue), respectively. (c) The heat map of the top 100 DEGs, the upregulated genes are shown in red, and the down regulated genes are shown in blue. Sample clustering result showed a good fit for the actual grouping method.
DEGs function exploration
DAVID provides a comprehensive set of functional annotation tools to understand biological meaning behind large list of genes. In this study, we used GO process to explore DEGs functions from three aspects, including cellular components, molecular function, and biological process. The results showed that DEGs were widely distributed in the whole cell and the extracellular regions, including exosomes and endocytic vesicles. Besides, the DEGs have a tight relationship with macrophage apoptosis, ion transport, inflammatory response, and cell differentiation, which also have an ability to bind proteins and function as cytokine or growth factor activators (Table 1). On the contrary, in order to understand the DEGs action mode, we adopted KEGG analysis, which was based on clustering similar genes into a same network, interestingly, the main action modes were cytokine–cytokine receptor interaction and transcriptional misregulation, the related signaling pathways were phosphoinositide 3-kinase (PI3K)-Akt, tumor necrosis factor (TNF), mitogen-activated protein kinase (MAPK), nuclear factor kappa B (NF-κB), RIG-I-like receptor, and Toll-like receptor (Table 2), which were consistent with previous studies in lung cancer.14–16
Gene ontology analysis of differentially expressed genes related to nano-silica treated macrophages.

GO: gene ontology.
KEGG pathway enrichment analysis of differentially expressed genes related to nano-silica treated macrophages.
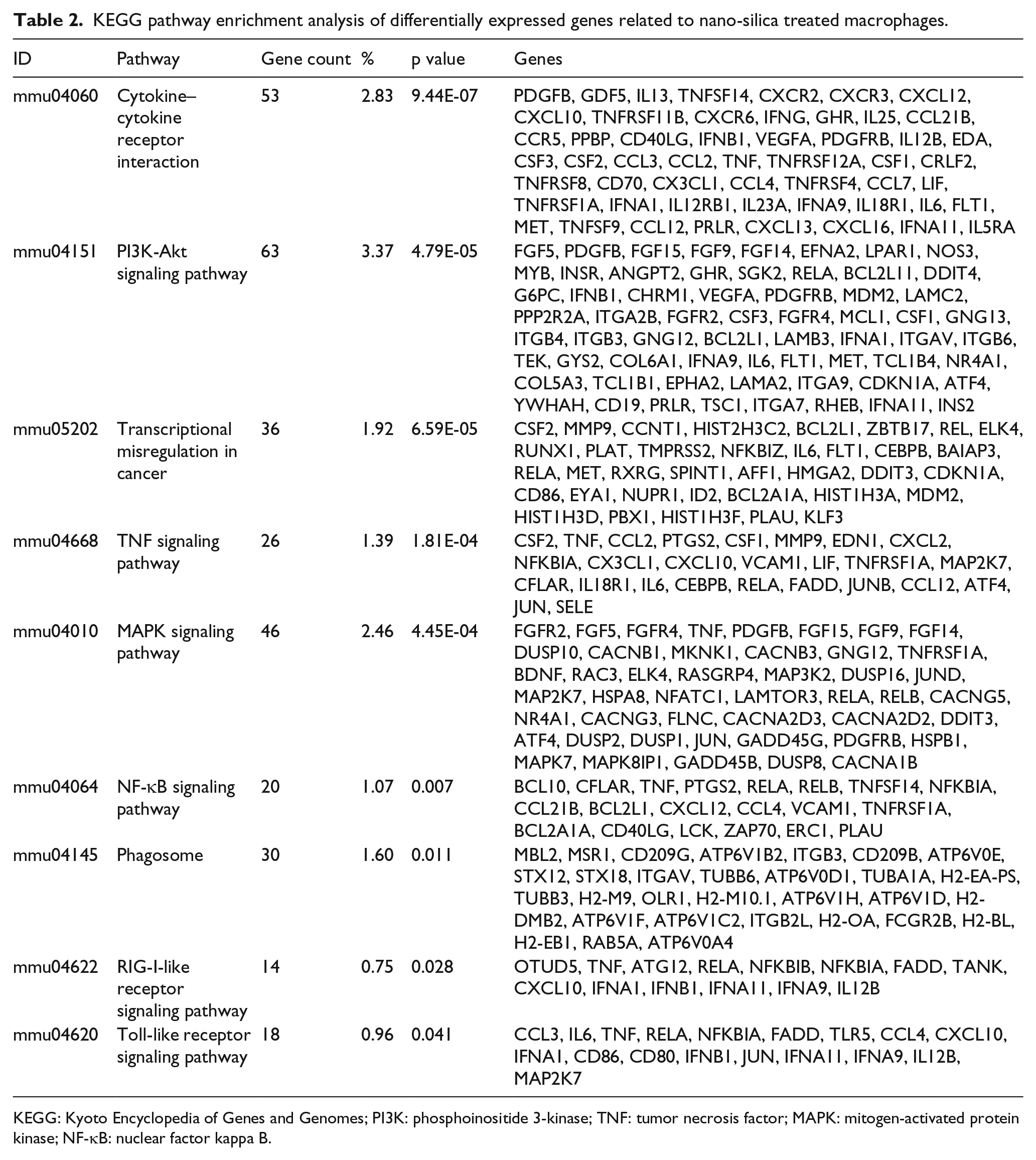
KEGG: Kyoto Encyclopedia of Genes and Genomes; PI3K: phosphoinositide 3-kinase; TNF: tumor necrosis factor; MAPK: mitogen-activated protein kinase; NF-κB: nuclear factor kappa B.
The PPI network construction
The PPI database was filtered using combined score >0.4, the top 10 hub genes, S100a9, Nos3, Psmd14, Psmd4, Lck, Atp6v1h, Jun, Foxh1, Pex14, and Fadd were identified from PPI network. Subsequently, records, which satisfied the filter criteria, were imported into Cytoscape for remodeling. Modules M1, M2, and M3, with a clustering coefficient ≥0.5, are shown in Table 3. The follow-up KEGG analysis pointed out that module M1 played an important role in neuroactive ligand–receptor interaction, cytokine–cytokine receptor interaction, calcium signaling pathway, and chemokine signaling pathway; module M2 mainly involved in the biosynthesis of proteasome and steroid, transcriptional misregulation, signaling pathways of TNF and hypoxia-inducible factor 1 (HIF-1); and module M3 had a close relationship with cell adhesion, arachidonic acid metabolism, PI3K-Akt, and Ras signaling pathways (Table 4 and Figure 2).
Module clustering of differentially expressed genes.

Pathways and differentially expressed genes in protein–protein interaction modules.
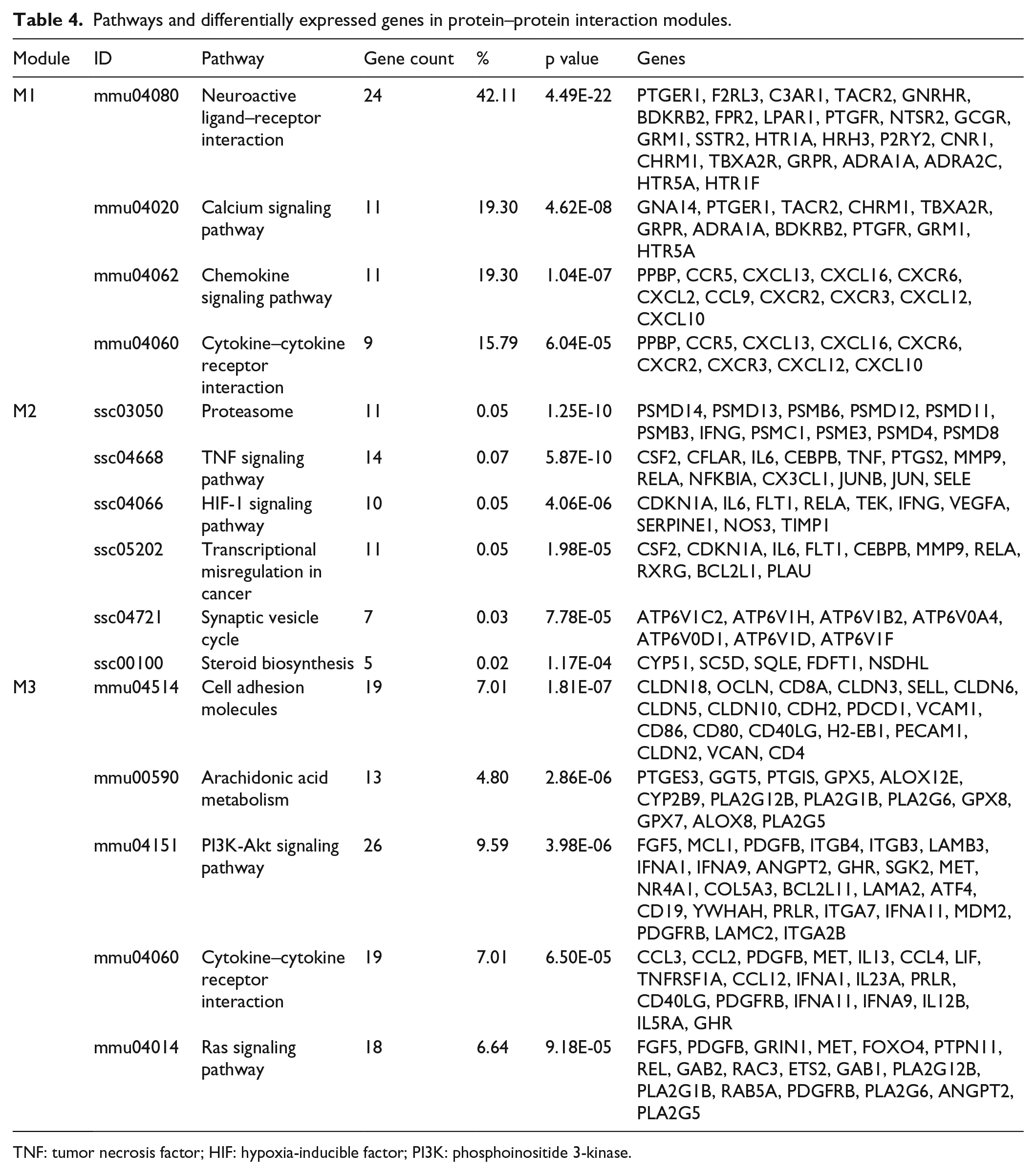
TNF: tumor necrosis factor; HIF: hypoxia-inducible factor; PI3K: phosphoinositide 3-kinase.

The module M2 protein–protein interaction (PPI) network. The PPI database was filtered using combined score >0.4, then records, which satisfied the filter criteria, were imported into Cytoscape for remodeling. According to the preset screening criteria (haircut mode, node score cutoff value 0.2, and K-core value 2), three modules were returned, of which, M2 contains the largest number of hub genes (red).
Discussion
Long-term inhalation of productive dust may cause pulmonary fibrosis, 17 which plays an important role in pneumoconiosis and lung cancer development. Since there is no effective treatment, the incidence of nanoparticle related disease has been increasing rapidly. As shown in many studies and reports, the number of new pneumoconiosis had reached 26,873 in 2014 in China, 9 besides, over 3 million workers exposed to silica dust in India, 18 about 2 million people have been working in dust exposure environment in America, of whom, the estimated death number of silicosis was 2500–5000 per year. 19 Moreover, in the World Health Report of 2002, World Health Organization (WHO) estimated that 30,000 deaths and nearly 1,288,000 disability adjusted life years (DALYs) occurred yearly due to exposure to occupational airborne particulates. 20 Fortunately, current studies have revealed that the development of pneumoconiosis is particularly associated with alveolar inflammatory cell infiltration. 21 Larson-Casey indicated that v-akt murine thymoma viral oncogene homolog 1 (Akt1) kinase–mediated mitophagy contributed to AM apoptosis resistance, which was required for pulmonary fibrosis. 22 Other studies suggested that sustained PI3K activation exacerbated lung fibrosis via activating pro-inflammatory and pro-fibrotic pathways, 23 but gene level evidence was insufficient. Similarly, AKT1-PI3K signaling pathway was proposed as a critical part in the apoptosis of macrophage which was treated with nano-silica, it was found that 63 genes were differentially expressed, but most of them were seldom studied in this field. For example, Nos3, a crucial member of the AKT1-PI3K signaling pathway, was mainly discussed in cardiovascular or urinary system diseases but rarely in pulmonary fibrosis. GO analysis suggested that Nos3 is widely distributed in the nucleus, cellular membrane, Golgi, mitochondrion, and vesicles, which also participates in many biological metabolism processes, including macrophage apoptosis, endothelial cell migration, cell proliferation, signal transduction, nitric oxide biosynthesis, removal of superoxide radicals, and lung development. Therefore, it would be of great significance to explore the role of Nos3 in pulmonary inflammation and fibrosis, and Nos3 may also become a therapeutic target in pneumoconiosis in a long-term perspective.
As is known to all, nanoparticle is one of the most important risk factors for lung cancer, which was proposed in the 1980s, and subsequently confirmed as sufficient evidence in humans for the carcinogenicity by the International Agency for Research on Cancer (IARC) in 1996. 20 Turci held that the carcinogenesis of quartz dust did not depend on crystallinity but conchoidal crystal fragmentation, which could induce cellular membrane disruption and toxicity, resulting in macrophage inflammatory response. 24 In addition, the oxidized macrophage migration inhibitory factor (MIF) was found to be a promoter of cancer or tumor via AKT–extracellular signal–regulated protein kinases 1 and 2 (ERK1/2) signaling pathway.25,26 Lim et al. 27 indicated that macrophages involved in pulmonary adenocarcinoma promoted tumor cell invasion and migration by upregulating the expression level of S100A8 and S100A9, which also accompanied by downregulating the matrix metalloproteinase 2 (MMP2) and MMP9. As a crucial component of DEGs, S100a8 and S100a9 are mainly distributed in the area of the plasma membrane, extracellular exosomes, and other regions, which have a capablility of binding metal ion, microtubules, and receptors. Moreover, S100a8 and S100a9 also take part in macrophage autophagy, apoptosis, leukocyte migration, inflammatory response, and innate immune response; however, both of them were not clustered in any models in this study, and the role of them still needs further mining and verification.
In addition to causing acute and chronic pulmonary diseases, nano-silica also induces autoimmune or autoimmune-related disorders, including scleroderma, systemic lupus erythematosus, rheumatoid arthritis, autoimmune hemolytic anemia, and dermatomyositis. Reports from the National Institute for Occupational Safety and Health (NIOSH) noted that there was an association between crystalline silica exposure with chronic renal diseases and sub-clinical renal changes. 20 Guo et al. 28 demonstrated an elevated messenger RNA (mRNA) level of collagen I and fibronectin in the heart and kidney after silica inhalation, but the severity of inflammatory response alleviated after anti-IL1β mAb treatment. In this study, we clustered the inflammatory response related signaling pathways in Table 2 and found that interleukin (IL13) might play an important role in macrophage response to silica stimulation. Interestingly, IL13 was also found to be an inducing factor in macrophage polarization in previous studies.29–31
As an important member of Psmd families, Psmd14 participates in the synthesis and secretion of proteasome, but excessive proteasome induces extracellular matrix deposition. 32 Atp6v1h, located in the lysosomal membrane and extracellular exosomes, was mainly involved in adenosine triphosphate (ATP) hydrolysis, synaptic vesicle cycle, and macrophage phagocytosis; however, available literatures for Atp6v1h only focused on its role in type 2 diabetes.33,34 Jun exists in the nucleus as a proto-oncogene, regulating cell apoptosis and the activity of the transcriptional enzyme. 35 Although the molecular functions of the above hub genes are generally clear, their roles in nanoparticle exposure related diseases are seldom reported. In this study, most of the hub genes were integrated into module M2, and the M2 clustering coefficient was 0.74, showing a good fitting effect and suggesting M2 of great significance in disease mechanism exploration.
In summary, DEGs screened from this study may provide new insights into the exploration of mechanisms, biomarkers, and therapeutic targets for diseases related to nanoparticle exposure, but subsequent studies are still needed to validate the hypothesis.
Footnotes
Acknowledgements
L.Z. and C.H. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Natural Science Foundation of China (81273039 and 81472954).