
Abstract
A1CF (apobec-1 complementation factor) acts as a component of the apolipoprotein-B messenger RNA editing complex. Previous researches mainly focused on its post-transcriptional cytidine to uridine RNA editing. However, few study reported its role in progression of breast carcinoma cells. Wound healing assay and flow cytometry were applied to detect the migration and apoptosis; western blot, real-time polymerase chain reaction, and dual-luciferase assays were applied to investigate the potential regulation mechanism of A1CF-mediated cell migration and apoptosis. Knockdown of A1CF decreased cell migration and enhanced cell apoptosis in MCF7 cells in vitro. Western blot analysis showed that knockdown of A1CF decreased Dickkopf1 but increased c-Myc and β-catenin expression, and overexpression of A1CF can get opposite results. Knockdown of Dickkopf1 in A1CF-overexpressed cells decreased cell migration and enhanced cell apoptosis compared with A1CF-overexpressed cells. Luciferase-fused 3′ untranslated region of human Dickkopf1 activity was highly upregulated in A1CF-overexpressed MCF7 cells, but this upregulation can be inhibited by mutating conserved binding motifs of Dickkopf1 3′ untranslated region. A1CF played a crucial role in cell migration and survival through affecting 3′ untranslated region of Dickkopf1 to upregulate its expression in MCF7 cells.
Introduction
Cell migration and apoptosis play crucial roles in tumor metastasis. Cell migration is essential for invasion and dissemination from primary solid tumors and for the establishment of lethal secondary metastases at distant organs. 1 Dysregulation in cell migration will increase the possibility of tumor cells to escape from the primary tumors. 2 Apoptosis, cell’s intrinsic death program, is an evolutionary highly conserved cellular process. If cell escapes from apoptosis, especially under the condition of limited nutrition, cell survival ability will be increased and finally the probability of cancer cells metastasis will also be increased in patients.3,4 Therefore, inhibiting migration or reactivating apoptosis of cancer cells is an effective way to treat cancer in the future, and finding new genes which function in cell migration and apoptosis is very helpful for expanding cancer therapeutic target sites.
Apobec-1 complementation factor (A1CF) belongs to heterogeneous nuclear ribonucleoproteins (hnRNP) family and is highly conserved across species.5,6 It contains three non-identical RNA recognition motifs (RRM) and a unique C-terminal auxiliary domain. 7 A1CF participates in the post-transcriptional cytidine (C) to uridine (U) RNA editing of apolipoprotein-B (ApoB) messenger RNA (mRNA) by co-acting with APOBEC1. 8 In addition, A1CF also regulates gene expression by controlling the stability of mRNA, as well as RNA editing. For example, A1CF modulates liver regeneration by binding to interleukin-6 (IL-6) mRNA 3′ untranslated region (UTR) to functionally strengthen IL-6 mRNA stability. 9 Besides, deletion of the homozygous of A1CF can lead to embryonic lethality due to the growth defect of blastocyst during preimplantation. 10 Therefore, participating in RNA editing and post-transcriptional RNA stability regulation may be the main ways for A1CF to function in various essential cellular processes, such as cell proliferation, cell apoptosis, cell migration, and cell differentiation. However, how A1CF regulates these processes remained unclear, especially in tumor cells.
Dickkopf1 (DKK1), a secreted protein which was an important inhibitor of Wnt/β-catenin signaling via blocking formation of Wnt-Frizzled-lipoprotein receptor-related protein-5/6 (LRP5/6) complex.11,12 Majority of studies had shown that DKK1 was involved in tumor biology. However, previous data also displayed controversial function of DKK1 in tumor cell migration and invasion. It might function as oncoprotein or tumor suppressor which is depended on cell types and their context.13–17
Here, we demonstrated that A1CF promoted cell migration and decreased cell apoptosis through upregulation of DKK1 expression in MCF7 cells, and the 3′UTR of DKK1 is a potential target region for A1CF to regulate DKK1 expression.
Materials and methods
Plasmid construction
Mouse A1CF CDS was obtained by polymerase chain reaction (PCR) and inserted into pCMV-flag vector at site of Xhol with the primers F: TCGACGGTATCGATAATGGAATCAAATCACAAATCC and R: GCTCTAGAACTAG TGAGGTTC CATATGCATCG. Homo sapiens A1CF short hairpin RNA (shRNA) was cloned into the pLKO.1 vector at sites of EcoRI and AgeI with shRNA sense: CCGGCCATGCTGCAAGGAGAGTATACTCGAGTATAC TCTCCTTGCAGCATGGTTTTTG and antisense: AATTCAAAAACCATGCTGCAAGGAGAGTAT ACTCGAGATACTCTCCTTGCAGCATGG. Homo sapiens DKK1 shRNA was cloned into the pLKO.1 vector at sites of EcoRI and AgeI with shRNA sense: CCGGGCCAGTAATTCTTCTAGGCTTCT CGAGAAGCCTAGAAGATTACTGGCTTTTTG and antisense: AATTCAAAAAGCCAGTAATTC TTCTAGGCTTCCGAGAAGCCTAGAAGAATTACTGGC. Homo sapiens DKK1 3′UTR was obtained by PCR from 293T cells complementary DNA (cDNA). Total RNA of 293T cells (treated with 40 mM lithium chloride for 24 h) was extracted by TRIzol reagent (Life Technologies, Grand Island, NY, USA), 18 and reverse transcription was performed by RevertAid First Strand cDNA Synthesis kit (Thermo Scientific, Walham, MA, USA) at the instance of manufacturer’s instructions. Homo sapiens DKK1 3′UTR was cloned into the pCDNA3.1-luciferase vector at sites of BamHI and EcoRI (pCDNA3.1-luciferase-DKK1-3′UTR-WT) with the primers F: CGCGGATCCACCAGCTATCCAAATGCAGTGAAC and R: CCGGAATTCAGGTATTATTAATTTATTGGAAACTATTTTTGAAAG. PCDNA3.1-luciferase-DKK1-3′UTR-MUT was acquired by introducing six mutations into the conserved ATTTA motifs of DKK1 3′UTR with the primers F1: GATAGTTTTTGAAATAAAACTGCACATTTAATATCA and R1: TTTCATGATATTAAATGTGCAGTTTTATTTCAAAAA, primer F2: GAAATAA AACTGCACGTCTGATATCATGAAATG and R2: AACATTTCATGATATCAGACGTGCG AGTTTTATT. A schematic diagram of the luciferase reporter constructs is shown in Figure 4(a).
Cell culture and transfection
Human embryonic kidney 293T (HEK293T) cells, MCF7 cells, and SKBR3 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS, vol/vol, Gibco) and 0.1% penicillin/streptomycin (15140122; Invitrogen, Grand Island, NY, USA), respectively. All the cell lines were cultured at 37°C with 5% CO2. For some experiments, cells were transiently transfected with plasmids using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. Then cell proteins were extracted at different time points and stored at −80°C for subsequent analyses.
RNA isolation and real time-PCR
PCMV-NC or pCMV-A1CF plasmids were transfected into MCF7 cells. After 48 h, the total RNA was isolated from MCF7 cells using Trizol (Life Technologies). We used RevertAid First Strand cDNA Synthesis kit (Thermo Scientific) for cDNA synthesis. Real time-PCR was performed using SYBR-Green qRT-PCR kit (ComWin Biotech, Beijing, China) according to the manufacturer’s instructions (Promega, Madison, WI, USA)—A1CF real-time PCR sense primer: TGCCTTCGTGGAGTATGAGA and antisense primer: CTCTGCCCAGTCTACTGC AA, DKK1 real-time PCR sense primer: CAGACTGTGCCTCAGGATTGTG and antisense primer: TGGCTTGATGGTGATCTTTCTG. The levels of A1CF mRNA and DKK1 mRNA expression were normalized with a housekeeping control gene (18s)—18s real-time PCR sense primer: GTAACC CGTTGAACCCCATT and antisense primer: CCATCCAATCGGTAGTAGCG.
Protein extraction and western blot
Western blot analysis was performed as previously described. 12 Cell proteins were lysed in lysis buffer with protease inhibitors at 4°C. Cell lysates were also collected and protein concentrations were determined using the BCA Kit (SK3051; Sangon, Shanghai, China). We used 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gel and polyvinylidene fluoride (PVDF) membrane (Millipore, Billerica, MA, USA) for western blot. Primary antibodies A1CF (1:2000, ab99955; abcam), DKK1 (1:2000, Proteintech, Wuhan, China), c-Myc (1:1000; Proteintech), β-catenin (1:1000, Proteintech), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:2000, Proteintech) were utilized after PVDF membrane was blocked in 5% fat-free milk (A600669; Sangon). And secondary antibody (1:3000; Proteintech) was utilized after PVDF membrane was washed with Tris-buffered saline and Tween 20 (TBST). Finally, we used the western blot chemiluminescent horseradish peroxidase (HRP) substrate (Immobilon Western, Billerica, MA, USA) to detect target proteins according to the manufacturer’s instructions. Internal control, GAPDH, was used to normalize the protein expression levels.
Wound healing assay
For some studies, when MCF7 cells confluence reached about 50%, pCMV-NC, pCMV-A1CF, pLKO.1-NC, pLKO.1-A1CF, or pLKO.1-DKK1 plasmids were introduced into MCF7 cells. We acquired time lapse images at different time points (12 and 24 h) with a fluorescence microscope (Nikon, Tokyo, Japan), and we collected images using a SPOT Diagnostic (Sterling Heights, MI, USA) CCD camera. We took photos in three independent selected fields of each sample, and the wound areas were calculated by Image J software (National Institutes of Health, Bethesda, MD, USA).
Transwell assay
To confirm the effect of A1CF on MCF7 cells migration, we transferred pCMV-A1CF and pLKO.1-NC/pLKO.1-DKK1 plasmids to MCF7 cells. Empty vector was set as control. A total of 1 × 105 MCF7 cells diluted in DMEM without FBS were plated into transwell chamber (Millipore) which was plated in 24-well plate containing DMEM with 10% FBS. After 12 h, cells that migrated to the bottom of the membrane were fixed with methanol and stained with crystal violet (Beyotime, Nanjing, China). Five fields were captured randomly under a microscope at 20×. Cell number of each group was normalized to the number of control to evaluate migration percentage. Experiment was performed triple times with three repeats in each group.
Cell apoptosis detection
For some studies, MCF7 cells were transfected with pCMV-NC, pCMV-A1CF, pLKO.1-NC, pLKO.1-A1CF, or pLKO.1-DKK1 plasmids for 48 h. Cell apoptosis was assessed using the Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (KGA107, KeyGEN, Nanjing, China) in accordance with the manufacturer’s instructions. Then cell apoptosis was determined by flow cytometry. The coordinate areas of Q2 and Q4 represent apoptotic cells in Figures 1 and 3.

Knockdown of A1CF decreases cell migration and promotes apoptosis in MCF7 cells. (a) Knockdown of A1CF significantly inhibited the migration ability of MCF7 cells and overexpression of A1CF can rescue cell migration defects in A1CF-depleted MCF7 cells. (b) Knockdown of A1CF significantly promoted apoptosis of MCF7 cells and overexpression of A1CF can inhibit cell apoptosis augments in A1CF-depleted MCF7 cells. (c) Migration data were collected at 12 and 24 h (original magnification, ×100) which displayed the same trend and represented mean ± SEM for three independent selected fields in each sample. (d) Statistical analysis of cell apoptosis was presented as mean ± SEM of three independent experiments in (b). (e) The silencing efficiency of A1CF-shRNA at protein level was detected by western blot, and GAPDH was applied to an internal control.
Dual-luciferase assays
For dual-luciferase assays, MCF7 cells were cultured into a 24-well plate at a density of 5 × 105 cells per well the day before transfection. Then the cells were transfected with 200 ng pCDNA3.1-luciferase-DKK1-3′UTR-WT or pCDNA3.1-luciferase-DKK1-3′UTR-MUT and 800 ng pCMV-NC or pCMV-A1CF for 48 h. A total of 10 ng of pRL-SV40 was included to serve as internal control. The luciferase activity was detected by the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions.
Statistical analysis
All experiments were performed at least in triplicates. The results were presented as the mean ± standard error of the mean (SEM). Student’s test was applied to test differences by statistical software Prism 5 (GraphPad, SanDiego, CA, USA), and significant differences were indicated as *p < 0.05, **p < 0.01, or ***p < 0.001.
Results
Knockdown of A1CF decreased cell migration and promoted apoptosis in MCF7 cells
To reveal the functions of A1CF in cell migration and apoptosis, wound healing assay and flow cytometry were applied to detect the migration ability and apoptosis rates of A1CF-depleted MCF7 cells. Knockdown of A1CF by specific shRNA decreased cell migration and promoted apoptosis in MCF7 cells (Figure 1(a) and (b)). Wound healing results of different time points (12 and 24 h) displayed the same trend (Figure 1(a)). Quantification analysis showed that cell migration was significantly inhibited in A1CF-depleted cells compared to control cells (Figure 1(c)), and overexpression of A1CF can rescue cell migration defects and apoptosis augments which were resulted by knockdown of A1CF in MCF7 cells (Figure 1(a) and (b)). These results indicated that A1CF played a critical role in cell migration and apoptosis. A1CF is downregulated after shRNA transfection or upregulated after overexpression by western blot analysis (Figure 1(e)).
A1CF promoted DKK1 expression but downregulated β-catenin and c-Myc in MCF7 cells
It is reported that DKK1 binding protein RBM47, a novel RNA-binding protein, has similar function with A1CF in APOBEC1-mediated RNA editing, 19 and RBM47 can suppress basal-like breast cancer cells progression and metastasis. 20 By sequence and structure comparison, we found that A1CF has high homology with RBM47 in conserved domain (Figure 2(a)) and three-dimensional (3D) structure (Figure 2(b)). Besides, RBM47 can stabilize DKK1 mRNA by binding to its 3′UTR. 20 These implied that A1CF may share similar mechanism with RBM47 to regulate the expression of DKK1. Therefore, we investigated whether A1CF also functions in DKK1 and DKK1-related Wnt signaling pathway. Our western blot results demonstrated that expression of DKK1 was decreased in A1CF-depleted cells compared with control cells (Figure 2(c) and (d)). Furthermore, some classic regulators in Wnt signaling pathway, such as β-catenin and c-Myc, were increased in A1CF-depleted MCF7 cells (Figure 2(c) and (d)), and overexpression of A1CF can get opposite results (Figure 2(e) and (f)).
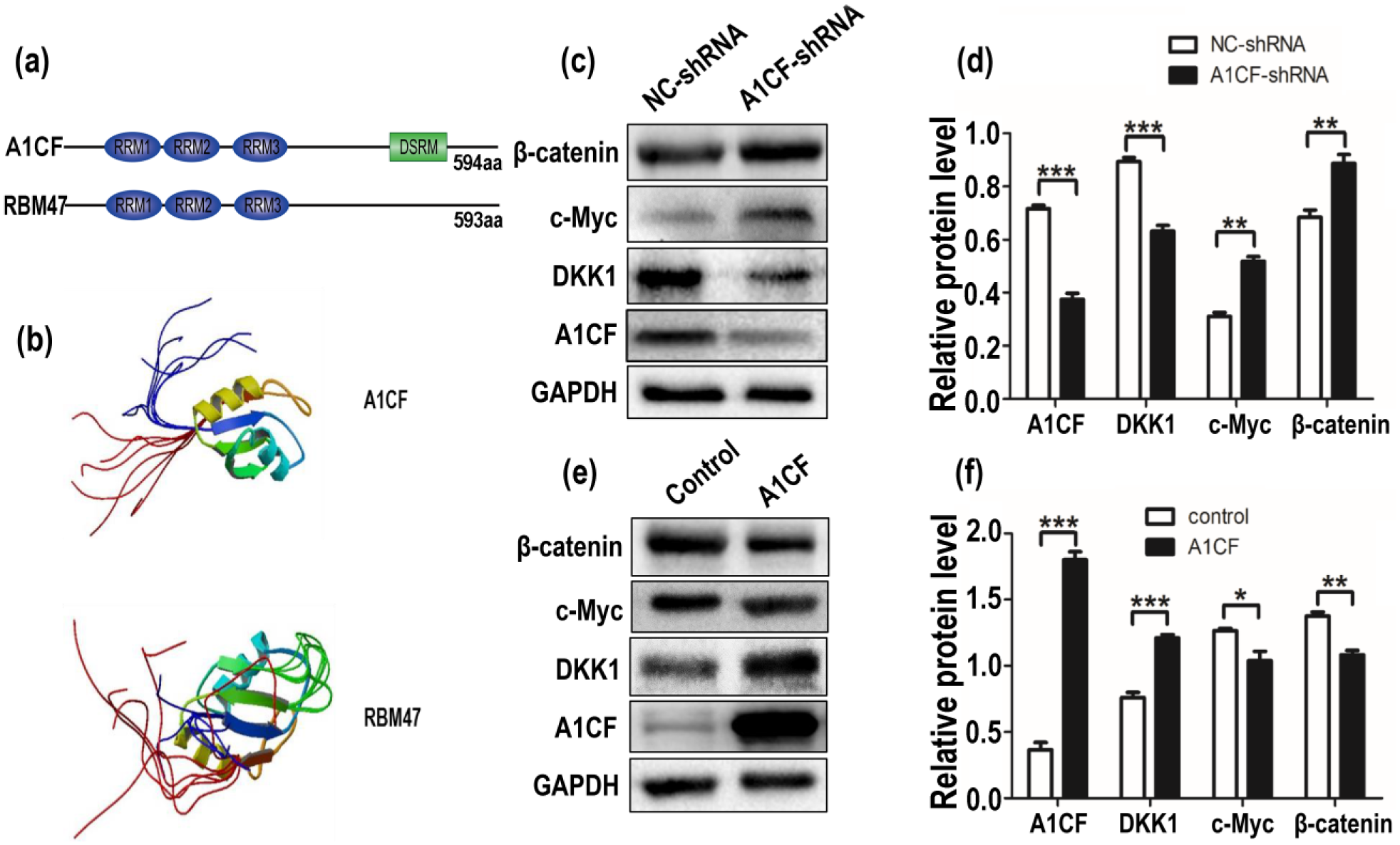
A1CF promotes the expression of DKK1 and further inhibits the expression of β-catenin and c-Myc. (a) Simulative protein structures of A1CF and RBM47 were online. A1CF and RBM47 both contain three non-identical RNA recognition motifs (RRM) on the N-terminus, and DSRM of A1CF represents double strand RNA-binding motif. (b) The solution of three-dimensional protein structures of A1CF and RBM47 was collected from the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do). (c) Western blot showed knockdown of A1CF downregulated DKK1 protein expression and further promoted the protein expression of β-catenin and c-Myc. (d and f) Quantitative analysis of A1CF, DKK1, c-Myc, and β-catenin protein levels after normalization with GAPDH. Data represented mean ± SEM for three independent experiments. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 indicate striking statistical differences in comparison to the control. (e) Western blot showed overexpression of A1CF upregulated DKK1 protein expression and inhibited β-catenin and c-Myc protein expression. GAPDH served as all loading control.
DKK1 was a necessary regulator of A1CF-mediated cell migration and apoptosis
To further validate the role of DKK1 in A1CF-mediated cell migration and apoptosis, we silenced DKK1 by specific shRNA in A1CF-overexpressed MCF7 cells, and wound healing assay and flow cytometry were employed to detect the phenotypes. Overexpression of A1CF increased cell migration and decreased apoptosis in MCF7 cells (Figure 3(a)–(c)). Wound healing results of different time points (12 and 24 h) displayed the same trend (Figure 3(c)). Similar results also obtained from Transwell assay (Figure 3(b)). Importantly, knockdown of DKK1 in A1CF-overexpressed cells decreased cell migration and increased apoptosis compared with A1CF-overexpressed MCF7 cells (Figure 3(a)–(c)). And DKK1-knockdown in MCF7 cells without A1CF expression showed decreased migration (Supplemental Figure). These data showed that DKK1 was involved in A1CF-mediated cell migration and apoptosis. Both overexpression and silence efficiencies were detected by western blot and GAPDH was served as an internal control (Figure 3(g)).

Knockdown of DKK1 inhibits the cell migration augments and rescues the cell apoptosis led by overexpression of A1CF in MCF7 cells. (a and b) Wound healing and Transwell assay showed knockdown of DKK1 inhibited the migration defects led by A1CF overexpression, negative and overexpression of A1CF in MCF7 cells served as control groups. Wound healing data were collected at 12 and 24 h (original magnification, ×100). Images of Transwell assay were captured at 12 h. (c) Knockdown of DKK1 rescued the cell apoptosis defects led by overexpression of A1CF in MCF7 cells. (d and e) Migration data were collected at 12 and 24 h which displayed the same trend and represented mean ± SEM for three independent selected fields in each sample. (f) Statistical analysis of cell apoptosis was presented as mean ± SEM of three independent experiments. (g) The overexpressing efficiency of A1CF and silencing efficiency of DKK1 shRNA at protein level were detected by western blot, and GAPDH was applied to an internal control.
UTR of DKK1 is a key region for A1CF to regulate DKK1 expression
To explore how A1CF regulates the expression of DKK1, we used the dual-luciferase assays and quantitative real-time PCR to detect the effect of A1CF on DKK1 3′UTR. It is reported that A1CF binds to AU-rich motif of IL-6 mRNA 3′UTR, especially AUUUA motifs, 9 and those motifs (AUUUA) are also existence on the 3′UTR of DKK1 mRNA, so we cloned 3′UTR of DKK1 and fused them with luciferase reporter gene (pcDNA3.1-luciferase), and we further substituted those motifs with ACATG and GTCTG (Figure 4(a)). And those replacements could break its regulation function. 9 These constructs were transfected into MCF7 cells along with pCMV-A1CF plasmids or negative control plasmids (pCMV-NC), and luciferase activity was detected by a dual-luciferase system. Luciferase activity was increased in A1CF-overexpressed MCF cells compared with the negative control, and mutant DKK1 3′UTR had little promotion to luciferase activity after overexpression of A1CF (Figure 4(b)). Real-time PCR results also demonstrated that DKK1 mRNA was significantly upregulated after overexpression of A1CF in MCF7 cells (Figure 4(c)). These results suggested that AUUUA motifs on DKK1 3′UTR played a crucial role in A1CF-increased DKK1 expression. In addition, we confirmed the results in other cell line, SKBR3. Western blot results showed that knockdown of A1CF decreased DKK1 expression, but increased the expression of c-Myc and β-catenin in SKBR3 cells (Figure 4(d) and (e)). Those results implied A1CF-increased DKK1 expression is not decided by cell type.

A1CF binds to DKK1 3′UTR and increases the expression of DKK1 mRNA. (a) We cloned 3′UTR of DKK1 (WT DKK1 3′UTR) into a luciferase reporter vector (pcDNA3.1-luciferase) and further mutated the conserved ATTTA motifs of DKK1 3′UTR (MUT DKK1 3′UTR). (b) WT DKK1 3′UTR and MUT DKK1 3′UTR reporters were transfected into MCF7 cells along with pCMV-A1CF plasmid or a negative control plasmid (pCMV-NC). Luciferase activity was enhanced in the overexpression of A1CF group compared with the negative control and mutating DKK1 3′UTR could partly inhibit the luciferase activity augments resulted by A1CF overexpression. (c) Compared with the negative control, DKK1 mRNA was significantly upregulated after transfection with pCMV-A1CF plasmids in MCF7 cells. These results demonstrate that A1CF can bond to DKK1 3′UTR and A1CF can increase the expression of DKK1 mRNA. (d) A1CF regulated the expression of DKK1 in another breast cancer cell line, SKBR3 cells. Western blot results showed that expression of DKK1 was decreased in A1CF-depleted SKBR3 cells compared with control cells, and expression of c-Myc and β-catenin was increased. (e) Quantitative analysis of A1CF, DKK1, c-Myc, and β-catenin protein levels after normalization with GAPDH. Data represented mean ± SEM for three independent experiments. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001 indicate striking statistical differences in comparison to the control.
Discussion
A1CF was identified as a componentery factor of the apoB mRNA editing complex. 7 Previous studies mainly focused on its post-transcriptional RNA editing (cytidine (C) to uridine (U)).21,22 Here, we demonstrated that A1CF played a crucial role in the migration and apoptosis of MCF7 cells in vitro (Figure 1). DKK1, a classic inhibitor of Wnt signaling pathway, was a key effector of A1CF in those processes (Figures 2 and 3). Luciferase reporter system further provided evidences that conserved motifs (AUUUA) of DKK1 mRNA 3′UTR are crucial for A1CF to regulate DKK1 mRNA level (Figure 4). These findings implied that A1CF is a potential oncogene and may function in cancer cell metastasis and apoptosis via DKK1.
A1CF is a component of the RNA editing complex that physiologically deaminates C to U on the mRNA of human lipoprotein ApoB.19,21,22 This conversion will produce premature termination of the protein and disrupt or enhance the function of target proteins. A1CF also binds to the 3′UTR of IL-6 mRNA and functionally strengthens the stability of IL-6 mRNA. 9 Our results suggested that A1CF was a positive regulator of DKK1 (Figures 2(c), (e) and 4) and finally affected the expression of other regulators in Wnt signal pathway, such as β-catenin and c-Myc (Figure 2(c) and (e)). 23 Besides, we got the similar results in another cell line, SKBR3; this implied that A1CF regulates DKK1 expression, which does not depend on cell type (Figure 4(d)).
We found that knockdown of A1CF inhibited cell migration and enhanced cell apoptosis in MCF7 cells (Figure 1). There is no reference about A1CF in cell migration and apoptosis before, but its effector protein, DKK1, is reported to promote hepatocellular carcinoma cell migration in human renal carcinoma cells; 13 our results are in good consistent with their reports. In this study, we tried to answer how A1CF regulates cell migration and apoptosis by acting on DKK1. Knockdown of DKK1 inhibited cell migration augments and rescued the cell apoptosis that resulted by A1CF overexpression (Figure 3(d) and (e)). The conserved motifs (AUUUA) of 3′UTR of DKK1 mRNA are potential target regions in A1CF-controled DKK1 expression. Those similar conserved motifs are also existing on 3′UTR of IL-6 mRNA that can be regulated by A1CF. 9 Based on these reports and our findings, we can conclude that A1CF may regulate DKK1 mRNA expression by acting on its 3′UTR.
In conclusion, A1CF increases cell migration and inhibits cell apoptosis through regulation of DKK1 mRNA expression, and conserved motifs (AUUUA) of 3′UTR of DKK1 are potential target regions in A1CF-controled DKK1 expression.
Footnotes
Acknowledgements
The authors thank the ScienLab team, an entrepreneur team of undergraduate students, for their help of plasmid vectors constructing. X.Y. and Q.L. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of China (Grant No. 31271563 and Grant No. 81572076) to Prof. Qin Zhou, MD, PhD, and grant from the National Basic Research Program of China (No. 2011CB944002) to Prof. Qin Zhou, MD, PhD.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.