
Abstract
The cancer germline antigens MAGE-A1, MAGE-A3, and NY-ESO-1 can be used to target relapsed or therapy-resistant malignant solid tumors, and previous studies have demonstrated that these antigens can be epigenetically upregulated on the surface of tumor cells following exposure to low-dose demethylating chemotherapy agents, such as decitabine. The extent to which cancer germline antigen cytotoxic T lymphocytes can be reliably expanded from healthy donors has not been well characterized, specifically in terms of whether these T cells consistently kill antigen-bearing targets or simply produce interferon-γ in the presence of the antigen. Cancer germline antigen cytotoxic T lymphocytes were generated using conventional method and high-density lymphocyte culture method. We demonstrate that there is no difference in the extent of antigen-specific killing with or without CD25 depletion when interleukin-21 is added to the cultures. Cancer germline antigen–specific killer cells could be expanded from 8/12 healthy donors using overlapping peptide mixes derived from MAGE-A1, MAGE-A3, and NY-ESO-1 and from 7/9 healthy donors using HLA-restricted epitopes. Furthermore, cytotoxic T lymphocyte derived from 4/5 patients displayed specific cytotoxicity of target cells expressing respective cancer germline antigen and HLA partially matched tumor lines. High-density lymphocyte culture prior to stimulation with cancer germline antigen peptides resulted in antigen-specific cytotoxic T lymphocyte from healthy donors and patients from whom cancer germline antigen cytotoxic T lymphocyte culture with conventional methods was not feasible. These data demonstrate that MAGE-A1-, MAGE-A3-, and NY-ESO-1-specific T cells with antigen-specific cytotoxicity can be cultured from healthy donors and patient-derived cells making adoptive immunotherapy with these cytotoxic T lymphocyte feasible.
Introduction
The cancer germline antigens (CGAs) MAGE-A1, MAGE-A3, and NY-ESO-1 have a restricted pattern of expression, limited to male germline cells, placenta, as well as a number of malignant solid tumors and leukemias, making these antigens of interest for immunotherapy.1–5 CGA expression can be modified epigenetically, and demethylating agents such as decitabine (5-aza-2′-deoxycytidine (DAC)) have been shown to upregulate their expression in a number of tumor cell lines,6,7 thereby facilitating their recognition by MAGE-A1-, MAGE-A3-, and NY-ESO-1-specific cytotoxic T lymphocyte (CTL).8,9
Several cancer vaccine studies have been conducted in adults and children with mixed clinical and immunological responses.10–12 The majority of patients receiving vaccines targeting CGA have failed to achieve sustained clinical responses, which could be due to problems developing primary immune responses to these antigens, as a result of immunocompromised status of these patients as well as the negative effects of regulatory T cells (Treg). 13 Since it can be difficult for patients with relapsed cancer to develop immune responses to these antigens, new strategies are needed to generate CGA CTL. Treg limits host immune responses to cancer, and increasing evidence suggests that Treg depletion can enhance antigen-specific immune responses to cancer vaccines.14,15 While Treg expresses CD25 and Foxp3, depletion of Treg using CD25 microbeads may eliminate activated antigen-specific cells. Subsequent studies have shown that interleukin-21 (IL-21) mediates Foxp3 suppression, and culturing T cells with IL-21 following Treg depletion leads to enhanced generation of antigen-specific CTL.16,17 Furthermore, IL-21 has been shown to play a significant role in the generation of primary antigen–specific CTL responses. 18
The precursor frequencies of CGA CTL are low to non-existent in many patients and healthy donors, and expanding these CTL is more laborious than the culture of CTL specific for viral antigens, such as cytomegalovirus (CMV) and Epstein–Barr virus (EBV). Therefore, conventional methods of CTL culture may need to be modified for the successful expansion of these CTL. Tumor antigen–specific CTL can be generated by cloning,19,20 sequential stimulation of T cells using antigen-pulsed autologous dendritic cells (DC), and by selection of activated cells based on interferon-γ (IFN-γ) secretion 21 or CD137 expression.22,23 Once tumor antigen-specific CTLs have been generated in vitro, there is no well-defined eligibility requirements or criteria to qualify these CTLs for adoptive immunotherapy. It is not known whether antigen-specific T cells that make cytokines in response to an antigen are also capable of killing tumor cells bearing these target antigens, particularly after these CTLs are infused for cancer immunotherapy. Previous studies have demonstrated that a dichotomy can exist between cytokine production and antigen-specific cytotoxicity.24,25 Although IFN-γ production and cytotoxicity are hallmarks of an effector CD8+ T cell, whether CGA-specific T cells should be qualified based on cytokine production alone is in question. We therefore developed an approach to culture CGA-specific CTL using MAGE-A1, MAGE-A3, and NY-ESO-1 overlapping peptide mixes or HLA-restricted epitopes. We show that CD25 depletion is not necessary when IL-21 is added to cultures for the generation of CGA CTL from healthy donors and that not all CGA CTLs that produce IFN-γ exhibit antigen-specific cytotoxicity and vice versa.
Materials and methods
Blood samples
Peripheral blood from patients and healthy adult donors was acquired after obtaining consent under a University of Louisville Institutional Review Board (IRB)-approved study.
Cell lines
Human NB cell lines CHP134 (DSMZ) and SKN-SH (American Type Culture Collection (ATCC)) were grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco-BRL) supplemented with 10% fetal bovine serum (FBS; Gemini). IMR32 (ATCC) was grown in RPMI (Gibco-BRL) supplemented with 10% FBS. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. Cells were counted and plated in T75 flasks 1 day prior to treatment, and then, the media was removed and replaced with fresh media containing 1 µM of DAC (Sigma-Aldrich) and/or 100 ng/mL human recombinant IFN-γ (R&D Systems) as indicated. After incubation with DAC and/or IFN-γ for 5 days, the cells were harvested, counted, and taken for functional assays.
Generation of DCs
DCs were generated as described previously. 10 Briefly, peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Paque (Amersham Biosciences) density gradient centrifugation from healthy adult donors. The monocytes were isolated by adherence of PBMCs and were cultured in CellGenix™ DC medium supplemented with human recombinant granulocyte-macrophage colony-stimulating factor (GM-CSF; 1000 U/mL; Sanofi) and interleukin-4 (IL-4; 10 ng/mL; R&D Systems). DCs were matured for 48 h in the presence of 10 ng/mL of tumor necrosis factor-α (TNF-α), 10 ng/mL of IL-1β, 10 ng/mL of IL-6 (R&D Systems), and 1 µg/mL of PGE2 (Sigma). Mature DCs were pulsed for 4 h at room temperature with MAGE-A1, MAGE-A3, and NY-ESO-1 overlapping peptide mixes (1 µg/mL of each peptide in peptide mix) consisting of pooled, 11-amino acid overlapping 15mers derived from the full-length protein (JPT Peptide Technologies) or previously identified immunodominant MAGE-A1, MAGE-A3, and NY-ESO-1 epitope mixes (1 µg/mL of each epitope in epitope mix; Mimotope) in the presence of 3 µg/mL β2-microglobulin. All investigational protocols were approved by the University of Louisville IRB.
Expansion of CGA-specific CTL
CTL generation by conventional method
Monocyte-depleted peripheral blood lymphocytes (PBLs) from healthy donors without or with CD25 depletion (using CD25 microbeads and passing them through a mini magnetic-activated cell sorting (MACS) column from Miltenyi Biotec) were stimulated with MAGE-A1, MAGE-A3, and NY-ESO-1 peptide pulsed autologous mature DC, at a T cell:DC ratio of 10:1. T-cell cultures suspended in CTL medium consisting of Click’s media (Sigma), RPMI 1640 (Gibco),
CTL generation by high-density pre-culture method
CGA CTLs were generated using the high-density pre-culture (RESTORE) protocol as described by Wegner et al. 26 with slight modification. Briefly, PBMC from healthy donors was placed in high density (107 PBMC/mL, 1.5 mL per well in 24-well plates) and cultured for 48 h. The pre-cultured T cells were stimulated every 7–10 days for 2–5 times with DC pulsed with CGA peptides, in the same manner as conventional CGA CTL. T-cell cultures (1 × 106 cells/well, 2 mL per well) were established in 24-well plates using CTL medium supplemented with 30 ng/mL IL-21 at a T cell:DC ratio of 10:1 and re-stimulated an additional two to five times (weekly) with IL-21 in culture and DC expressing these CGA at the same PBL:DC ratios. From the time of the second stimulation, the pre-cultured CTLs were further supplemented with 50 IU/mL IL-2, 10 ng/mL IL-7, and 10 ng/mL IL-15 (Miltenyi Biotec) 1 day after the stimulation.
Cytotoxicity assay
Chromium release assay
Cytotoxicity was determined by labeling the targets with 51Cr (PerkinElmer), as previously described. 9 CTLs were added at a responder:target ratio of 25:1. After 4 h of incubation, the supernatant was analyzed in a gamma counter. Spontaneous and total release for each target was used to calculate percentage-specific release by the following formula: % specific release = (experimental cpm − spontaneous cpm)/(total cpm − spontaneous cpm). A positive response is defined as ≥15% killing and twice of the control. Cytotoxicity was determined by chromium release assay unless otherwise specified.
Non-radioactive cytotoxicity assay
Cytotoxicity was determined by Cytotox 96 non-radioactive cytotoxicity assay kit (Promega) as per the instructions from the manufacturer. Briefly, CTLs were added to target cells at a ratio of 25:1 in a 96-well plate (total 100 µL volume). After 4 h of incubation at 37°C, 50 µL of cultured supernatants from each well was transferred to a flat-bottom 96-well plate. Equal volume (50 µL) of substrate from the kit was added to the culture supernatant, and the reaction was stopped after 30 min. Absorbance was read on Thermo Fisher MULTISKAN GO spectrophotometer at 490 nm wavelength. Percentage cytotoxicity was calculated from the formula: % cytotoxicity = ((experimental − effector spontaneous − target spontaneous)/(target maximum − target spontaneous)) × 100.
Cytokine production by ELISPOT
The resulting CTLs were characterized for specific IFN-γ production in response to stimulation with target cells described above at weekly intervals, using the BD ELISPOT Set (BD Biosciences). CTLs were cultured with target cells at a ratio of 5:1 and incubated overnight. Spots were developed according to the instructions from the manufacturer and counted using an automated ImmunoSpot Analyzer (CTL—Cellular Technology Ltd) as previously described. 9 Each group was done in triplicates and the average was taken. The number of spots in each group was calculated by subtracting the number of spots in control wells (with CTL only) from the number of spots in each group (CTL and target cells). A positive response is defined as >200 spots/million cells and twice of the control. In antibody-blocking studies, the targets were incubated with 10 µg/mL of relevant mAb for 1 h at 37°C before adding the CTL. The antibodies used include anti-HLA-A, B, C mAb (W6/32), anti-HLA-DR mAb (L243), and mouse IgG2a kappa isotype control (BioLegend).
Statistical analysis
Data analyzed by Student’s paired t test with p ≤ 0.05 were considered statistically significant.
Results
Bypassing CD25 depletion does not affect the cytotoxic efficacy of CGA-specific CTL
Here, we demonstrate that MAGE-A1-, MAGE-A3-, and NY-ESO-1-specific CTL can be generated from healthy donors following two to five weekly stimulations of T cells with autologous DCs pulsed with respective antigens and with IL-21 in culture. Tregs are a subset of T cells that have shown to control T-cell responses against viral and tumor antigens.27,28 CTLs were stimulated with DC loaded with either peptide mixes (pMix) derived from full-length NY-ESO-1, MAGE-A1, and MAGE-A3 or multiple HLA-restricted epitopes for each CGA (referred to as epitope mix). Selection of either form of peptide antigens to stimulate donor CTL was based on the HLA typing of each donor and the availability of epitopes matching with donor HLA (Supplementary Table 1). Li and Yee 16 reported that a combination of CD25 depletion (to reduce Treg) followed by the culture of monocyte-depleted lymphocytes with tumor antigens and IL-21 resulted in robust cancer antigen–specific T-cell responses. IL-21 is a gamma-chain receptor cytokine like IL-2, IL-7, and IL-15, which influences T-cell activation and differentiation and decreases the number of Treg.16,29 To determine whether CD25 depletion is necessary to expand CGA-specific CTL, we cultured PBL from four healthy donors with and without CD25 depletion and with the addition of IL-21 (Figure 1). CTL culture with the addition of IL-21 and without CD25 depletion resulted in antigen-specific cytotoxicity from each of these donors, with preferential lysis of DAC-treated, partially HLA-matched neuroblastoma cell lines. HLA match chart between donor and tumor line is given in supplementary Table 2. The MAGE-A1-specific response in donor 2 and the MAGE-A1- and MAGE-A3-specific response in donor 4 were significantly higher in the non-CD25-depleted group. Furthermore, CGA CTL generated from donor 2 and donor 4 without CD25 depletion demonstrated significant cytotoxicity of tumor lines treated with DAC and IFN-γ. In general, from four of these donors, there was no significant improvement in cytotoxicity in the CD25-depleted group when compared to non-depleted T cells, suggesting that removal of CD25-positive cells might not be necessary, as long as IL-21 is added to the culture. These data suggest that CGA-specific CTL can be cultured after at least three weekly stimulations when IL-21 is added to the culture, without the need for advanced cell processing.
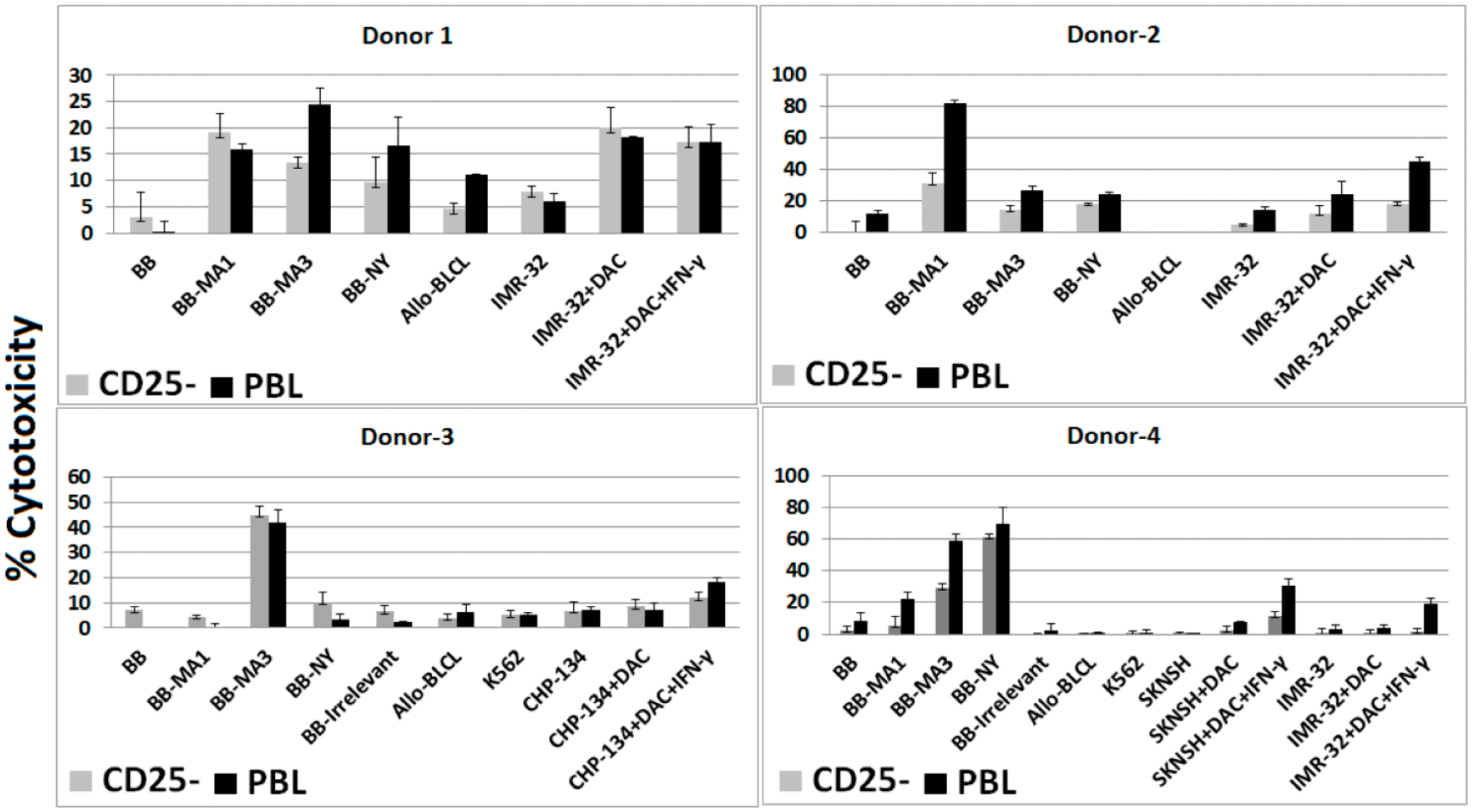
Cytotoxicity assay. CTLs were cultured from healthy adult donor PBL with and without CD25 depletion, and the cells were stimulated 2–5 times with MAGE-A1, MAGE-A3, and NY-ESO-1 peptide mix (donors 1–3) and epitope mix (donor 4). Error bars represent standard deviation of the measurements. Target cells include autologous B-cell blasts (BB), BB pulsed with peptides, allogeneic B-lymphoblastoid cell line (allo-BLCL), and HLA partially matched tumor cells with and without treatment with DAC and IFN-γ.
Feasibility of generating and expanding CGA-specific killer cells by conventional method using epitope mix and peptide mix
Previous studies have demonstrated that CGA-specific CTL generated by stimulating with immunodominant epitopes have efficient cytolysis and/or cytokine production.30–32 However, targeting tumor using single antigen may not be ideal due to single-antigen loss, a commonly seen tumor escape mechanism. In this study, we used multiple immunodominant epitopes of CGA (Supplementary Table 1), referred to as epitope mix (EpiMix) to expand the application to a wider patient population. EpiMix was used in CTL generation by stimulating cells from donors whose HLA typing partially matches at least one MHC Class I and one Class II antigen. Furthermore, to expand CTL against a wide range of epitopes irrespective of patient HLA typing, we used overlapping peptide mixes (pMix) derived from full-length MAGE-A1, MAGE-A3, and NY-ESO-1 to characterize cytotoxicity and cytokine (IFN-γ) production by CTL. Therefore, to determine whether pMix or EpiMix could enhance the feasibility of expanding CGA-specific CTL, we stimulated 13 healthy donor T cells (PBL without CD25 depletion) with pMix- or EpiMix-pulsed autologous DC by conventional method (as described in “Materials and methods” section; Table 1). Four of nine (44%) conventional CTL generated using pMix and five of seven (71%) EpiMix CTL displayed efficient lysis of autologous B blasts (BB) expressing specific antigens (Figure 2(a)). CTL stimulated with pMix or EpiMix recognized multiple antigens. Analysis of cytotoxicity after stimulation with pMix or EpiMix showed recognition of either one (donors 2, 3, and 5), two (donors 1 and 6), or all three (donors 4, 9, and 11) of the CGA.
Cytotoxicity and cytokine production by conventional CTL.

ND: not determined; CTL: cytotoxic T lymphocyte.

(a) Cytotoxicity by conventional CGA CTL. Cytolysis of target cell by CTL generated using pMix (upper panel) and EpiMix (lower panel; () BB; ( ) BB-MA1; (
) BB-MA1; ( ) BB-MA3; (
) BB-MA3; ( ) BB-NY; (
) BB-NY; ( ) BB-Irr. Error bars represent standard deviation of the measurements and *p ≤ 0.05). (b) Cytokine production by conventional CGA CTL. IFN-γ production by CTL generated using pMix (upper panel) and EpiMix (lower panel; () BB; () BB-MA1; () BB-MA3; () BB-NY; () BB-Irr. Error bars represent standard deviation of the measurements and *p ≤ 0.05).
) BB-Irr. Error bars represent standard deviation of the measurements and *p ≤ 0.05). (b) Cytokine production by conventional CGA CTL. IFN-γ production by CTL generated using pMix (upper panel) and EpiMix (lower panel; () BB; () BB-MA1; () BB-MA3; () BB-NY; () BB-Irr. Error bars represent standard deviation of the measurements and *p ≤ 0.05).
Cytokine production by conventional pMix and EpiMix CGA-specific cells and their functional heterogeneity
Although it is well established that effector cells, especially CD8+ T cells, produce cytokines, chemokines, and lyse target cells expressing specific antigens, it is still unclear whether in vitro stimulated pMix and EpiMix CGA-specific cells are functionally homogeneous with regard to cytokine production and cytolysis. To address this question, we analyzed the production of IFN-γ by conventional pMix and EpiMix CGA-specific cells using ELISPOT and examined their ability to kill target cells (Table 1). ELISPOT and cytotoxicity assays were performed at the same time using effector cells that were expanded after two to five stimulations. pMix-stimulated T cells from four of seven (57%) donors and EpiMix-stimulated cells from five of seven (71%) donors showed CGA-specific IFN-γ production (Figure 2(b)).
It is assumed that effector cells can produce cytokines, especially IFN-γ, and also kill target cells. However, during differentiation, antigen-specific cells undergo functional segregation. Cytokine production and cytotoxicity are regarded as two distinct functions of CD8+ effector cells. To assess the functional properties of in vitro generated CGA-specific conventional effector cells stimulated using pMix or EpiMix, we examined the cytolytic activity and IFN-γ production (Table 1). Four of seven (57%) pMix-stimulated CTL demonstrated functional differences, three donor cells produced IFN-γ after two to three stimulations, but no lytic activity was seen after five rounds of weekly stimulation. In addition, one donor (donor 5) had cytolytic activity after two stimulations but did not produce IFN-γ after three stimulations. Similar to pMix CTL, two of seven (29%) EpiMix-stimulated CTL demonstrated functional differences in IFN-γ production and cytolysis. One of the two donors had cytolytic activity but did not produce IFN-γ and vice versa. Since some of these IFN-γ-producing CGA-specific effector cells were incapable of lysing antigen-specific targets, the qualification of effector cells solely based on IFN-γ production may not accurately define which CTL has the capacity to kill tumor cells. Another factor to be considered in our experimental system is that the CTLs are cultured from PBL (heterogeneous sample with both CD4 and CD8 T cells) and pMix could sensitize both class I and II epitopes. It is therefore possible that the differences in cytolysis and cytokines are due to the emergence of dominant CD4 populations and/or age of the T cells in these cultures.
High-density pre-culture method may enhance the effector functions of CGA-specific pMix and EpiMix CTL
Placing CTL in dense culture prior to antigenic stimulation has been shown to facilitate the acquisition of antigen-specific effector functions at earlier time points than control CTL. 26 We examined whether high-density pre-culture could enhance our ability to culture highly efficient CGA-specific CTL for cellular immunotherapy. We combined the high-density pre-culture method with IL-21 and other cytokines (IL-7, IL-15, and IL-2) to facilitate the expansion of CGA-specific CTL stimulated with DCs pulsed with MAGE-A1, MAGE-A3, and NY-ESO-1 peptides and studied IFN-γ production and CGA-specific killing (Table 2). Four of five (80%) pMix-stimulated CTL and 2/2 (100%) EpiMix CTL had cytolytic activity (Figure 3(a)). All CTL irrespective of stimulation with pMix or EpiMix produced IFN-γ (Figure 3(b)). One of five (20%) pMix CTL produced IFN-γ, but failed to demonstrate cytolytic activity even after five rounds of stimulation. Donor 1 and donor 8 pMix-stimulated effector cells did not have antigen-specific cytotoxicity after six stimulations when cultured using the conventional method (as mentioned in “Materials and methods” section), but had CGA-specific cytotoxicity after five stimulations and two stimulations, respectively, with high-density pre-culture. These results suggest that the high-density pre-culture method may enhance the feasibility of culturing CGA CTL that not only produce IFN-γ but also kill target cells in an antigen-specific manner.
Cytotoxicity and cytokine production by high-density pre-culture CTL.
ND: not determined.
(a) Cytotoxicity by high-density pre-culture CTL. Cytolysis of target cell displayed by CTL generated using pMix (upper panel) and EpiMix (lower panel; () BB; () BB-MA1; () BB-MA3; () BB-NY; () BB-Irr. Error bars represent standard deviation of the measurements and *p ≤ 0.05). (b) Cytokine production by high-density pre-culture CTL. IFN-γ production by CTL generated using pMix (upper panel) and EpiMix (lower panel; () BB; () BB-MA1; () BB-MA3; () BB-NY; () BB-Irr. Error bars represent standard deviation of the measurements and *p ≤ 0.05).
Both conventional and high-density pre-culture methods lead to a similar fold expansion of CTL. After five rounds of stimulation, CTL cultured by the conventional method resulted in a 3.5-fold expansion, while the high-density culture method was associated with a 3.8-fold expansion. We could not study the expansion of antigen-specific cells since MAGE-A1, MAGE-A3, and NY-ESO-1 CTLs were generated using peptide libraries for each antigen. However, Li and Yee 16 reported a 200-fold increase in antigen-specific CTL by combining CD25 depletion and IL-21 addition by peptide–MHC tetramer staining.
Finally, both peptide mixes and HLA-restricted epitopes could stimulate CGA-specific T cells using either the conventional or pre-culture methods. The number of CGA CTL that could both kill and produce IFN-γ was higher with epitope mix stimulation than with peptide mix; 8 of 12 (67%) pMix and 7 of 9 (77%) EpiMix-stimulated CTL demonstrated cytolysis, while 7 of 10 (70%) pMix and 7 of 9 (77%) EpiMix CTL produced IFN-γ. These results suggest that both pMix and EpiMix are efficient sources of antigen in stimulating CGA-specific CTL.
Generation of CGA-specific killer cells from neuroblastoma and sarcoma patients
CGA CTLs were cultured using high-density pre-culture method from three neuroblastoma (patients 1, 2, and 5), one rhabdomyosarcoma (patient 3), and one osteogenic sarcoma (patient 4) patient. Since pMix can be used to stimulate CGA CTL irrespective of their HLA typing, patient-derived T cells were stimulated with MAGE-A1, MAGE-A3, and NY-ESO-1 pMix-pulsed autologous DCs in the same manner as healthy donors. The resulting CTLs were analyzed for their ability to kill target cells. Due to the temporary unavailability of chromium within USA while patient-derived CTLs were ready to be assayed, we used the alternative non-radioactive cytotoxicity assay. As shown in Figure 4(a), the CTL displayed specific cytotoxicity against BB pulsed with respective CGA as well as against HLA partially matched tumor lines treated with DAC and IFN-γ. HLA match chart between patient and tumor line is given in Supplementary Table 2. Our group has previously reported that the expression of MAGE-A1, MAGE-A3, and NY-ESO-1 can be upregulated on neuroblastoma cells following exposure to DAC. Furthermore, we have shown that culture of neuroblastoma cell lines with IFN-γ was associated with an increased expression of MHC I and II molecules. 8 Patient-5 derived cells did not display cytotoxicity even after five rounds of stimulation (data not shown). Patient-2 pMix-stimulated effector cells did not have antigen-specific cytotoxicity after five stimulations when cultured using conventional method (data not shown), but did display CGA-specific cytotoxicity after five stimulations with high-density pre-culture method (Figure 4(a)).

(a) Killing of HLA partially matched tumor lines by patient-derived CGA CTL. The CGA CTL was stimulated with pMix and cultured by high-density pre-culture method. Error bars represent standard deviation of the measurements. Target cells include autologous B-cell blasts (BB), BB pulsed with peptides, and tumor cells with and without treatment with DAC and IFN-γ. (b) Cytokine production by patient-derived CGA CTL. IFN-γ production by CTL generated using pMix (upper panel). Antigen recognition by MAGE-A3 CTL was blocked using antibodies against HLA-A, B, and C (αMHC Class I) and HLA-DR (αMHC Class II) or an isotype control (Iso) antibody (lower panel). Blocking was performed using patient-3 MAGE-A3 CTL that had been previously cultured and cryopreserved.
While four of five (80%) pMix-stimulated patient-derived cells displayed cytotoxicity against targets bearing respective CGA, only two of five (40%) patient-derived CTL produced IFN-γ (Figure 4(b)). Pre-treatment of target cells with anti-HLA class I mAb significantly inhibited the production of IFN-γ by CGA CTL, whereas an anti-HLA class II mAb and the isotype control antibodies had no effect (Figure 4(b), lower panel). This clearly indicates the conventional HLA class I-restricted nature of CGA CTL.
Discussion
Cancer vaccines have been used in children and adults with relapsed malignant solid tumors for the past two decades, but with limited success due to several factors, including tumor escape mechanisms as well as tumor- and chemotherapy-induced immunosuppression. MAGE-A1, MAGE-A3, and NY-ESO-1 are highly immunogenic and have been targeted in numerous cancer vaccine trials.10,33–36 While CGA-directed immune responses have been reported in oncology patients receiving these vaccines, the magnitude of these responses is often low and often not associated with sustained clinical responses, possibly due to the level of immunocompromise in these patients.10,35,36 The likelihood of patients achieving a clinical and immunological response may be enhanced if ex vivo–expanded CGA CTL with known anti-tumor activity could be directly infused into these patients, bypassing the need for a primary immune response to these antigens after vaccination. To this end, we sought to optimize CGA CTL culture conditions and determine the feasibility of expanding CGA CTL from healthy donors. We were successful in culturing multi-CGA-specific CTL from healthy donors using a conventional method of stimulating CTL weekly with DCs pulsed with HLA-restricted epitope mixes or overlapping CGA peptides. Furthermore, we show that high-density pre-culture can enhance the ability to generate highly efficient CGA-specific CTL for cellular immunotherapy.
A challenge in culturing CGA CTL is the fact that Treg limits the ability of these CTL to expand in vivo and in vitro. 37 Previous studies have demonstrated that the negative effects of Treg can be mitigated by the use of cytokines such as IL-21 and Treg depletion. With IL-21 known to mediate Foxp3 suppression and also reduce Treg numbers, 16 we cultured MAGE-A1-, MAGE-A3-, and NY-ESO-1-stimulated PBL with IL-21, thereby bypassing CD25 depletion to further simplify the process of culturing CGA CTL for clinical use. Our results demonstrate that there was no apparent benefit on CTL cytotoxicity by depleting PBL of CD25+ cells, as all four donors had antigen-specific killing irrespective of Treg depletion. CD25 is not only a marker on Treg but also expressed on activated T cells. Furthermore, Treg expresses Foxp3 and previous studies have shown that IL-21 mediates Foxp3 suppression. This together with our results suggests that IL-21 could act through suppressing Foxp3, thereby facilitating the generation of CGA-specific CTL responses.
Furthermore, optimizing CGA CTL culture requires the identification of an antigen product that consistently stimulates CGA CTL. The choice of antigens includes whole protein, individual human leukocyte antigen (HLA)-restricted epitopes, and overlapping peptide mixes derived from each full-length CGA. Stimulation using both peptide mixes and HLA-restricted epitopes led to the induction and expansion of CGA-specific CTL from healthy donors. Results from our study indicate that HLA-restricted epitopes were most likely better in stimulating CGA CTL than peptide mixes. It should be noted, however, that we did not test generation of CGA CTL using peptide mix and epitope mix in all the donors and compare their function. There is very little data on antigen-specific cytotoxicity of peptide mix–stimulated CTL and whether native CGAs are recognized by CTL stimulated in this manner. 38 While overlapping peptide mixes permit the culture of CTL irrespective of a patient’s HLA background, it is possible that with peptide mixes, individual epitopes are cut and irrelevant epitopes could be presented.
Previous studies with EBV- and CMV-specific T cells have used cytotoxicity as the standard criteria for qualifying in vitro generated antigen-specific cells for adoptive immunotherapy.39–41 While other groups have demonstrated that CGA-reactive T cells can be cultured from healthy donors using peptide mixes, these T cells have been qualified as CGA specific primarily based on specific IFN-γ production. 42 In our study, not all IFN-γ producing CGA CTL cultured by the conventional method lysed target cells. Furthermore, it might not be ideal to select CGA-specific T cells based on IFN-γ production alone because in some donors, the effector cells produced IFN-γ after two to three stimulations, but gained cytotoxic ability only after four to five stimulations (data not shown). Conversely, 3/5 CGA CTL cultured from patients did not produce IFN-γ even after five stimulations. Therefore, it is essential that the in vitro generated CGA CTL should be qualified based on their cytolytic ability and not based exclusively on IFN-γ production.
Another challenge in culturing CGA CTL is the fact that the frequency of these cells in the peripheral blood is low to undetectable, requiring multiple cycles of stimulation, selection, or cloning, which can lead to T-cell exhaustion. Wegner et al. 26 demonstrated that viral- and WT1-specific CTL activity could be greatly enhanced by first pre-culturing lymphocytes in dense culture for 48 h prior to antigenic stimulation. This strategy helps CD4 cells regain antigen sensitivity (as exists in lymph nodes and is lost in the peripheral circulation) and re-establish cell contact, enhancing the ability of T cells to respond to antigenic stimuli. Our results demonstrate that culturing cells in high density, prior to stimulation by MAGE-A1-, MAGE-A3-, and NY-ESO-1-pulsed autologous DC could improve the feasibility of culturing CGA CTL from healthy donors. The high-density pre-culture method led to the generation of CGA CTL that is capable of killing antigen-specific target cell even in donors from whom CTL culture was not feasible by the conventional method.
Tumors undergo several escape mechanisms to prevent immune recognition. One such mechanism is downregulation of CGA expression on tumor cells, which could be reversed by epigenetic modulators.8,43 A combination of DAC followed by CGA CTL could enhance the recognition and killing of tumor cells expressing CGA.10,23 The appearance of antigen-loss variants is another common tumor strategy used to evade the immune system. Previous studies have shown that antigenic competition does not appear during CTL generation with multiple tumor antigens.35,44 The use of CGA CTL targeting multiple tumor antigens could overcome the limitation of selected loss of targeted antigens.33,35,45
Taken together, our results indicate that the combination of a high-density pre-culture and IL-21 added to the CTL leads to the generation of CGA-specific cytotoxic CTL from healthy donors and patients. MAGE-A1, MAGE-A3, and NY-ESO-1 are relevant antigens for immunotherapy since they are expressed on different types of adult and pediatric solid tumors and leukemia. Our strategy could be used to culture autologous CGA CTL from patients with solid tumors for cellular immunotherapy or CGA CTL derived from healthy donors could be used for adoptive therapy to prevent or treat relapse following allogeneic stem cell transplantation.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funds from RaiseRED and Hyundai Hope on Wheels.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.