
Abstract
Breast cancer is a malignant tumor that is harmful to women’s health around the world. Investigating the biological mechanism is, therefore, of pivotal importance to improve patients’ prognoses. Compared to non-neoplastic tissues, enhanced glucose and lipid metabolism is one of the most common properties of malignant breast cancer. Adenosine triphosphate (ATP) citrate lyase is a key enzyme linking aerobic glycolysis and fatty acid synthesis and is of high biological and prognostic significance in breast cancer. In our clinical study, fresh clinical tissues were used to analyze ATP citrate lyase expression by western blotting, and paraffin archived samples from 62 breast cancer patients were used to analyze ATP citrate lyase expression by immunohistochemistry. In the cellular study, following small interfering RNA–mediated inhibition of ATP citrate lyase in MCF-7 cells, cell viability and apoptosis were measured using the Cell Counting Kit-8 and flow cytometry, respectively. Breast cancer tissues showed strong expression of ATP citrate lyase, whereas adjacent normal tissues showed weak expression. Silencing of endogenous ATP citrate lyase expression by small interfering RNA in MCF-7 cells suppressed cell viability and increased cell apoptosis. Collectively, our study revealed that expression of ATP citrate lyase was significantly increased in breast cancer tissue compared with normal tissue. In addition, we found that depletion of ATP citrate lyase suppressed tumor growth, which suggests that ATP citrate lyase–related inhibitors might be potential therapeutic approaches for breast cancer.
Introduction
Breast cancer (BC) is a leading cause of cancer-related death for women around the world. 1 Increasing evidence indicates that cancer is not only a genetic disease but also a metabolic disease.2–5 Cancer cells reprogram their metabolism to promote growth, survival, proliferation, and long-term maintenance. The common feature of this altered metabolism is an increased rate of glucose uptake and lactate production, even in the presence of sufficient oxygen. This phenomenon was observed by Otto Warburg nearly 90 years ago and is now termed “the Warburg effect.” Among these pathways, fatty acid synthesis stands out as cancer cells prefer de novo lipogenesis over internalization and utilization of extracellular lipids regardless of lipid concentration in the extracellular matrix.6,7 Elevated levels of fatty acid synthesis are required during cancer cellular growth and proliferation. 8 Today, at least three enzymes in this process are known to play key roles in tumor progression, namely, ATP citrate lyase (ACLY), acetyl coenzyme A carboxylase (ACC), and fatty acid synthase (FAS).
ACLY converts glucose-derived citrate to cytosolic acetyl-CoA (acetyl coenzyme A) and oxaloacetic acid (OAA) in the presence of ATP and CoA 9 (Figure 1). OAA is decomposed into pyruvate by malic dehydrogenase and malic enzyme, which reenters into the next citrate pyruvate cycle. Meanwhile, ACC converts cytosolic acetyl-CoA to malonyl-CoA, which serves as a substrate of fatty acids. Cytosolic acetyl-CoA is required for several important biosynthetic pathways, including lipogenesis and cholesterogenesis. 10 ACLY is the key enzyme in catalyzing the first step of de novo lipogenesis 11 and linking aerobic glycolysis and fatty acid synthesis.12,13 Moreover, ACLY activity is indispensable to growth factor–induced nuclear histone acetylation and gene expression. 9 In addition, ACLY is vital to fetal growth and development. 14 Based on these findings, we assumed that ACLY upregulation is a process of cancer development. However, several studies have revealed that ACLY is highly expressed in various types of tumors but not in normal cells.15–17 Meanwhile, ACLY dysregulation contributes to metabolic alterations. Recently, researchers found that inactivation of ACLY suppressed cancer viability and inhibited cancer growth. 2

Brief schematic diagram representing the pathway of de novo lipogenesis. ACLY catalyzes the conversion of citrate to OAA and acetyl-CoA in the presence of ATP and coenzyme A. ACC catalyzes cytosolic acetyl-CoA to malonyl-CoA, and OAA decomposes to pyruvate, which reenters the next citrate pyruvate cycle. Malonyl-CoA is used as a substrate and catalyzed by FAS to form fatty acids.
Considering these findings, ACLY might be a potential biomarker and an effective therapeutic target for cancer. In this study, we examined the expression of ACLY in 62 pairs of BC and adjacent normal tissues and evaluated the impact of ACLY on BC cell growth, proliferation, and metastasis.
Materials and methods
Cell culture and reagents
The estrogen-sensitive human breast adenocarcinoma cell line MCF-7, the estrogen-independent adenocarcinoma cell line MDA-MB-231, and immortalized mammary epithelial cell line MCF-10A were obtained from XiangYa Central Experiment Laboratory (Changsha, China) and maintained in RPMI1640 medium (GIBCO, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin in a humidified incubator with 5% CO2 at 37°C.
Clinical samples
A total of 62 pairs of BC and adjacent normal tissue were obtained from patients who underwent surgical resection at the Second Affiliated Hospital of Soochow University between June 2013 and December 2014. All BC cases were histologically confirmed. This study was approved by the Ethics Committee of the Second Affiliated Hospital of Soochow University, and informed consent was obtained from all patients.
Immunohistochemistry
Tissue microarrays (TMAs) of 62 BC cases were constructed for immunohistochemistry (IHC). The samples were fixed in 10% neutral formalin, embedded in paraffin, and serially sectioned at 5 µm. TMAs were deparaffinized and rehydrated using xylene and graded ethanol. After deparaffinization and antigen retrieval, which was performed for 30 min in citrate buffer (pH 6.0), slides were blocked with 5% bovine serum albumin for 1 h and incubated overnight at 4°C in primary antibody (p-ATP citrate lyase (p-ACLY), 1:100, Abcam, Cambridge, MA, USA; anti-HER2/neu, Dako, Denmark; anti–estrogen receptor (ER), Dako; anti–progesterone receptor (PR), Dako; anti-Ki-67, Copenhagen, Dako). Sections were washed with phosphate-buffered saline (PBS) three times for 5 min and labeled with rabbit horseradish peroxidase (HRP)-conjugated second antibody for 20 min at 37°C. Visualization was performed with 3′-diaminobenzidine tetrahydrochloride (DAB) and counterstained by hematoxylin. The area of positive staining was scored on a scale of 0–4: 0, none; 1, poor; 2, moderate; 3, moderate to strong; 4, strong. The frequency of positive cells was defined as follows: 0, less than 5%; 1, 5%–25%; 2, 26%–50%; 3, 51%–75%; and 4, greater than 75%. When the staining was heterogeneous, we scored it as follows: each component was scored independently and summed for the results. For example, a specimen containing 75% tumor cells with moderate intensity (3 × 2 = 6) and another 25% tumor cells with weak intensity (1 × 1 = 1) received a final score of 6 + 1 = 7. For statistical analysis, scores of 0–7 were considered low expression and scores of 8–12 considered high expression.
ER/PR was considered positive if 1% or more of tumor cells have nuclear staining of any intensity according to the ASCO/CAP (American Society of Clinical Oncology/College of American Pathologists) guideline recommend. 18
HER2 was defined as positive if 10% or more of tumor cells exhibit strong uniform membrane staining according to the ASCO/CAP guideline recommend. 19
Ki-67 was regarded positive as the stained cell percentage was ⩾20% according to the 2013 St Gallen conference. 20
Synthesis of siRNAs
A double-stranded small interfering RNA (siRNA) oligonucleotide targeting ACLY (sense, 5′-UUCUUGAUCAGCUUUCUCGUGAGGG-3′; antisense, 5′-CCCUCACGAGAAAGCUGAUCAAGAA-3′) was designed and synthesized by Shanghai Genepharma Co. Ltd (Shanghai, China). Negative controls were designed by varying these sequences and were not homologous to any known sequences in GenBank (sense, 5′-UUCUCCGAACGUGUCACGUTT-3′; antisense, 5′-ACGUGACACGUUCGGAGAATT-3′). The siRNAs were dissolved in siRNA dilution buffer (Shanghai Genepharma Co. Ltd) to a final concentration of 20 µmol/L.
Transfection of siRNAs
MCF-7 cells (3 × 105) were seeded onto 35 mm six-well tissue culture plates and allowed to adhere for 24 h. Prior to transfection, 5 µL of Lipofectamine™ 2000 transfection reagent (Invitrogen, Carlsbad, CA, USA) and 5 µL of siRNA solution were added to Buffer EC-R (Qiagen, Hilden, Germany) to prepare a total volume of 500 µL per well. The complex was gently mixed, incubated at room temperature for 20 min, and added to wells containing 2 mL Dulbecco’s modified Eagle’s medium (DMEM) and 10% FBS; cells were then incubated under normal cell culture conditions. Untransfected control cells (UC group) and negative control cells (NC group) were prepared in parallel.
Western blotting
To obtain protein lysates, frozen tissues and cells were homogenized in lysis buffer (Beyotime, Shanghai, China) containing 8 M urea, 10% sodium dodecyl sulfate (SDS), 1 M dithiothreitol (DTT), protease inhibitors, and phosphatase inhibitors and centrifuged at 20,879g at 4°C. Total protein concentration was measured using the bicinchoninic acid (BCA) assay. Organic and cellular extracts containing 30 µg total protein were separated on 10% SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) gels and then transferred onto polyvinylidene difluoride membranes (Invitrogen). The membranes were incubated for 2 h in blocking solution containing 5% non-fat dry milk to inhibit non-specific binding and then incubated with primary anti-ACLY (1:1000, Abcam), anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:1000, Hangzhou Goodhere Biotechnology Co., Hangzhou, China), and anti-p-ACLY (1:1000, Abcam) antibodies at 4°C overnight. After washing with tris-buffered saline with Tween-20 (TBST) (0.5% Tween-20 in tris-buffered saline (TBS)), the membranes were incubated with HRP-conjugated secondary antibodies (1:4000, MultiSciences Biotech Co., Hangzhou, China) in TBS and 1% milk for 2 h at room temperature. The bands were visualized using enhanged-chemi-luminescence (ECL) (1:4000, MultiSciences Biotech Co.) and exposed to Kodak X-ray films for 30 s–2 min. Films were scanned and band intensities were determined using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Reverse transcription polymerase chain reaction
Total cellular RNA was isolated using TRIzol (Invitrogen) according to the manufacturer’s instructions. For reverse transcription polymerase chain reaction (RT-PCR), 5 µg of total RNA per sample was reverse transcribed using the Reverse Transcription Reaction Kit (Fermentas, St. Leon-Rot, Germany) according to the manufacturer’s instructions. Complementary DNA (cDNA) (1 µL) was amplified by polymerase chain reaction (PCR) (pre-denaturation step at 95°C for 5 min followed by 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s, and then 72°C for 10 min). The primers used were: ACLY, 5′-GAAGGGAGTGACCATCATCG-3′ and 5′-TTAAAGCACCCAGGCTTGAT-3′, GAPDH, 5′-TCCTGTGGCATCCACGAAACT-3′ and 5′-GAAGCATTTGCGGTGGACGAT-3′. The reaction products were visualized by electrophoresis on 1.5% agarose gels using 1× TBE (tris-borate electrophoresis) buffer containing 0.5 µg/mL ethidium bromide. The final normalized results were calculated by dividing the relative transcript levels of the target genes by the relative transcript levels of GAPDH.
Cell viability determination
Cell viability was tested using the CCK-8 (Cell Counting Kit-8, Dojindo Molecular Technologies, Rockville, MD, USA) according to the manufacturer’s instructions. Cells were seeded (2 × 103/well) in a 96-well flat-bottomed plate and treated with the indicated conditions at 37°C. After RNA interference group for 48 h, 20 µL of CCK-8 was added to each well, cells were incubated at 37°C for 0.5 h, and the optical density at 450 nm was measured using a microplate absorbance reader (M200; Tecan, Grodig, Austria). Cells that stained positively with the CCK-8 solution were considered viable, and cell viability was presented as percent.
Flow cytometry assay
For apoptosis analysis following RNA interference for 48 h, cells were washed, resuspended in ice-cold binding buffer, and incubated with 5 µL Annexin-V-fluorescein isothiocynate (FITC) (Beyotime) and 10 µL propidium iodide for 15 min. Cell apoptosis analyses were performed with a flow cytometer (FC500; Beckman Coulter, Brea, CA, USA). Each experiment was repeated in triplicate.
Statistical analysis
Statistical analysis was performed using SPSS 13.0 (SPSS, Chicago, IL, USA). All data are presented as mean ± standard deviation, and one-way analysis of variance (ANOVA) with Dunnett’s T3 post hoc test was used to determine statistical significance. Data from cell culture experiments were compared using Student’s t-tests. For clinical samples, correlations between p-ACLY expression and clinicopathological parameters were assessed using Fisher’s exact test. Univariate survival analysis was performed using the Kaplan–Meier method and log-rank statistics for comparisons of survival curves. Multivariate survival analysis was determined by the Cox regression model. Comparisons of ACLY expression between BC and normal tissue were carried out using the Mann–Whitney U test. Differences between groups were analyzed using two-tailed t-tests. The value of p < 0.05 was considered statistically significant.
Results
p-ACLY and ACLY expressions in BC and adjacent normal tissue
BC tissues showed strong expression of ACLY as well as p-ACLY, whereas adjacent normal tissues showed weak expression (Figure 2). The expression of ACLY in BC tissue was significantly higher than that in adjacent normal tissue (Figure 2(a) and (c), **p < 0.01). The expression level of p-ACLY in BC tissue was significantly higher than that in adjacent normal tissue (Figure 2(b) and (d), *p < 0.05).

Expression of ACLY and p-ACLY in normal and tumor breast tissues. Western blot was performed as described in section “Materials and methods.” (a, c) ACLY was higher in tumor than that in normal tissue (n = 4, **p < 0.01); (b, d) p-ACLY was higher in tumor than in normal tissue (n = 4, *p < 0.05); (c, d) statistical analysis of western blot results of a and b, respectively. The values of p were calculated by Student’s t-tests.
The relationship between p-ACLY expression and clinicopathological features of BC
Previous studies have indicated that phosphorylated ACLY is its biological active form. 21 Three phosphorylation sites have been identified on ACLY, namely, threonine 446, serine 450, and serine 454. Insulin and β-adrenergic receptor agonists induce an increase in serine 454 phosphorylation, the latter agonist acting directly via the activation of PKA (protein kinase A). 21 Additionally, phosphorylation enhances the stability of the protein. To investigate the relationship between p-ACLY expression and clinicopathological characteristics, we evaluated p-ACLY expression by IHC (Figure 3). High p-ACLY expression was related to tumor size (p = 0.02), pathology grade (p = 0.032), lymph node metastasis (p = 0.039), and Ki-67 expression level (p = 0.038). There were no significant associations between ACLY expression and age, ER, PR, or HER2 expression, or clinical pathological type (all ps > 0.05) (Table 1).
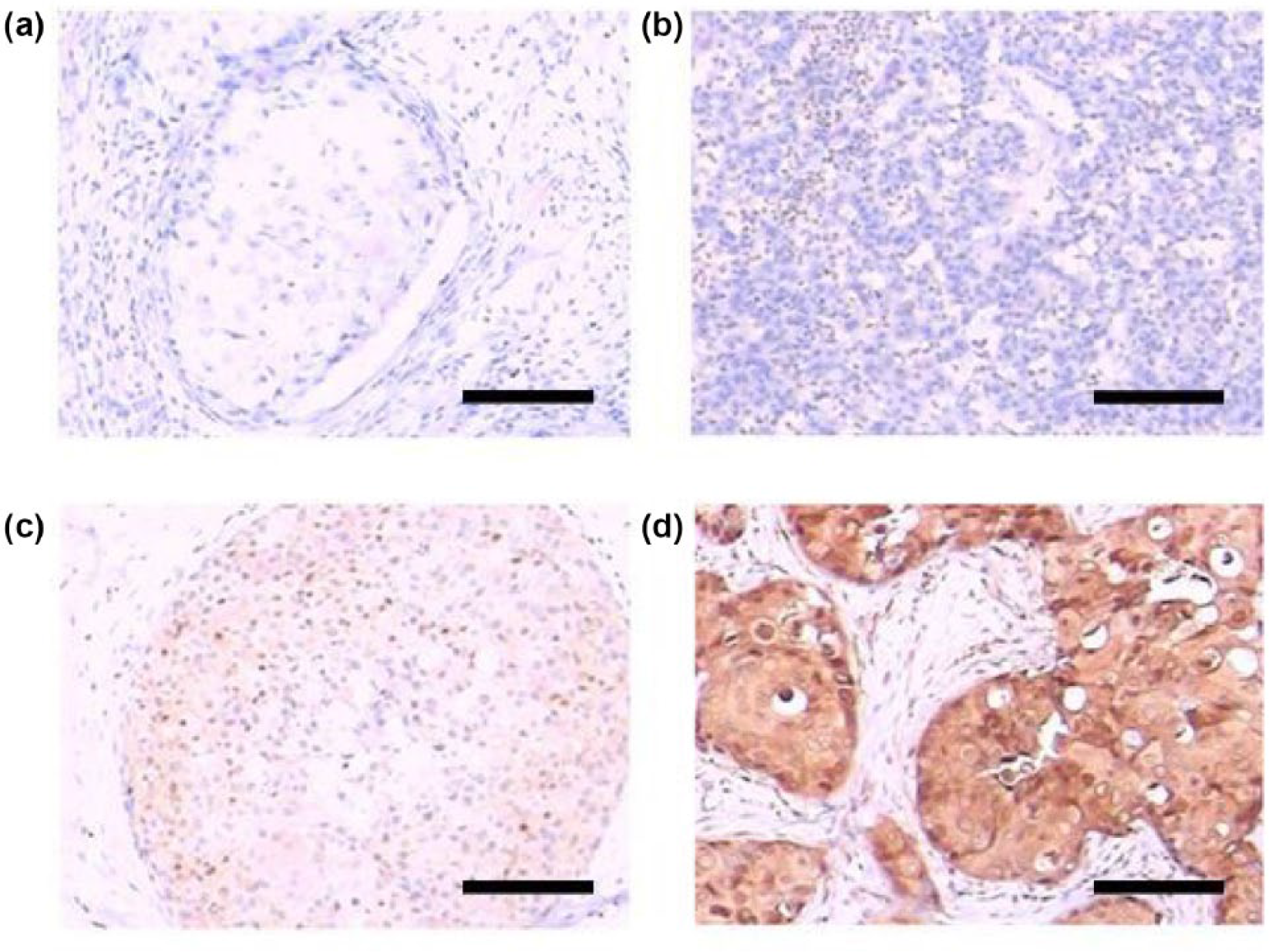
Immunohistochemical analysis of p-ACLY in BC tissues: (a) negative control (no primary antibody), (b) weak p-ACLY expression (0 score), (c) moderate p-ACLY expression (2 score), and (d) strong p-ACLY expression (4 score); original magnification: ×100, scale bar = 200 µm, original magnification (d): ×400, and scale bar (d) = 50 µm.
The relationship between clinicopathological characteristics and tumor p-ACLY expression levels of breast cancer patients.

p-ACLY: p-ATP citrate lyase; ER: estrogen receptor; PR: progesterone receptor.
ACLY and p-ACLY expressions in MCF-7, MCF-10A, and MDA-MB-231
ACLY and p-ACLY levels were higher in BC cell lines MCF-7 and MDA-MB-231 as compared to immortalized breast epithelial cell line MCF-10A (Figure 4). The expression of ACLY in MCF-7 cells and MDA-MB-231 was significantly higher than that in MCF-10A cells (Figure 4(a) and (c), *p < 0.05, **p < 0.01). The expression level of p-ACLY in MCF-7 cells and MDA-MB-231 was significantly higher than that in MCF-10A cells (Figure 4(b) and (d), *p < 0.05, *p < 0.05).

Western blot showing expression levels of ACLY and p-ACLY in different cell lines. Western blot was performed as described in section “Materials and methods.” (a, c) ACLY was significantly higher in MCF-7 cells than in MCF-10A cells (n = 4, *p < 0.05); ACLY was significantly higher in MDA-MB-231 cells than in MCF-10A (n = 4, **p < 0.01). (b, d) p-ACLY was significantly higher in MCF-7 cells than in MCF-10A cells (n = 4, *p < 0.05); p-ACLY was significantly higher in MDA-MB-231 cells than in MCF-10A (n = 4, *p < 0.05); the expression of ACLY and p-ACLY had no significant difference between MCF-7 cells and MDA-MB-231 cells. (c, d) Statistical analysis of western blot results of a and b, respectively. The values of p were calculated by Student’s t-tests.
Confirmation of ACLY silencing in MCF-7 cells by siRNA
To confirm RNA interference (RNAi)-mediated silencing of ACLY in MCF-7 cells, ACLY mRNA (messenger RNA) and protein expression were quantified by RT-PCR and western blotting, respectively. As shown in Figure 5(a), the PCR products of ACLY and GAPDH were 358 and 225 bp, respectively (no marker in Figure 5(a)). The expression of ACLY mRNA decreased compared to NC group (Figure 5(b), **p < 0.01); there were no significant differences between UC group and NC group. More importantly, ACLY protein level robustly decreased in siRNA-transfected MCF-7 cells compared to control cells (Figure 5(c) and (d)).

The levels of mRNA and protein expression of ACLY in MCF-7 cells. (a) RT-PCR analysis and (b) quantification of ACLY mRNA expression in MCF-7; mRNA levels were normalized to the loading control GAPDH. The ratio of ACLY to GAPDH mRNA was calculated for each group. Data are mean ± SD of three independent experiments; NC group: negative control cells; UC group: untransfected control cells; E: experimental group (siRNA ACLY); **p < 0.01 compared with NC; *p < 0.05 compared with UC. (c, d) Immunoblotting analysis and (e, f) quantification of ACLY (c, e) and p-ACLY (d, f) protein expression in MCF-7 cells. Blots were stripped and re-probed with GAPDH antibody to ensure equal protein loading. The ratio of ACLY and p-ACLY to GAPDH protein was calculated for each group. Values are mean ± SD of three independent experiments; NC group: negative control cells; UC group: untransfected control cells; E: experimental group; **p < 0.01 compared with NC; *p < 0.05 compared with UC.
Depletion of ACLY inhibits cell viability and promotes cell apoptosis
Next, we determined the effect of silencing ACLY on MCF-7 cell viability and apoptosis using the CCK-8 assay and flow cytometry, respectively. As shown in Figure 6, flow cytometry analysis demonstrated that treatment of MCF-7 cells with ACLY siRNA significantly increased cell apoptosis compared to the control group (*p < 0.05). Further experiments indicated that cell viability was reduced after ACLY depletion (Figure 6(e)).

Effect of ACLY on cell apoptosis and cell viability. (a–d) Apoptosis rates of cells detected by flow cytometry. Apoptosis of ACLY siRNA-transfected MCF-7 cells significantly increased compared to NC and UC groups; NC group: negative control cells; UC group: untransfected control cells; E: experimental group. (e) After 48 h of transfection, proliferation of MCF-7 cells in the ACLY siRNA-transfected group was determined by CCK-8 cell viability assay kit. *p < 0.05; NC group: negative control cells; UC group: untransfected control cells; E: experimental group.
Discussion
Malignant tumor cells need more energy metabolism to adapt to increased proliferation. A number of studies have indicated that endogenous fatty acid synthesis increased in human cancer cells, and that regulation of the fatty acid synthesis pathway is not affected in normal cells. 11 Abnormal lipid metabolism is involved in energy synthesis, growth, and even drug tolerance of tumor cells. In recent years, abnormal lipid metabolism of malignant tumors has become a hot topic of research in this field.
It has been well established that ACLY is the first key enzyme in the production of acetyl-CoA, which is needed for de novo lipogenesis as well as acetylation reactions.22,23 Overexpression or activation of ACLY has been previously found in cancers.12,16,24–27 In this study, we first observed elevated ACLY in BC tissue and its clinical significance by analysis of 62 different BC TMAs. Our data suggested that the expression of ACLY was associated with pathological grade, tumor size, Ki-67 expression level, and lymph node metastasis. Its expression was not related to the expression of ER, PR, and HER2 in BC. ER, PR, and HER2 are important prognostic factors for BC, and they are also important proteins in BC classification. HER2 (ERBB-2), a member of the epidermal growth factor (EGF) receptor family (also known as type I or ERBB receptor tyrosine kinases), 28 is overexpressed in about 30% of human breast tumors where it correlates with poor prognosis. 29 One major signaling pathway of the ERBB family is the RAS-RAF-MAPK (mitogen-activated protein kinase) pathway.30,31 Another important signaling route is PI3-kinase and its downstream protein kinase AKT/Protn Kinase B. 32 The latter signaling pathway stimulates lipogenic gene transcription through activation of the lipogenic transcription factor sterol regulatory element binding protein-1 (SREBF1) and directly activates lipogenic enzymes such as ACLY; this pathway links the upregulation of lipogenesis in cancer cells to the well-known tumor-associated increase in glycolysis.21,25,33 However, we found that the expression of ACLY was not related to HER2 expression, and this may suggest that ACLY can be phosphorylated by other kinases such as nucleoside diphosphate kinase 34 or cyclic adenosine monophosphate (AMP)-dependent protein kinase. 35
The ER is a 66-kDa nuclear protein and a member of the steroid hormone receptor superfamily. The presence of ER and progesterone receptors (PgR) in BC tissue is a reliable indicator of its hormone dependency. Furthermore, ER and PgR are important prognostic factors, and their presence is correlated with better clinical outcomes and lower risks of mortality.36–38
Ki-67 is a nuclear non-histone protein and is a proliferation marker expressed in the G1, S, G2, and M phases of the cell cycle. 39 Ki-67 is connected with cellular functions such as cell cycle regulation, ribosomal RNA processing, and DNA organization.39,40 Many previous studies have already demonstrated that Ki-67 expression is the strongest individual factor associated with poor differentiation of tumors and tumor size in BC.41,42 Numerous studies have also shown that high Ki-67 expression has a negative relationship with ER expression. 42 One study showed an inverse correlation between expression of the hormone receptors, ER and PR, and Ki-67. 42 This study indicated that Ki-67 was correlated with breast tumor size, pathological grade, and lymph node metastasis in 356 BC patients. In our study, ACLY expression was associated with pathological grade, tumor size, Ki-67 expression, and lymph node metastasis, but not with HER2, ER, or PR expression. The mechanism linking Ki-67 and ACLY is unknown; however, we can speculate from the above evidence that ACLY may be as useful a prognostic factor as Ki-67. The link between ACLY and additional clinicopathological parameters is worth examining more closely.
We determined the expression of ACLY and p-ACLY in estrogen-sensitive human breast adenocarcinoma cell line MCF-7, the estrogen-independent adenocarcinoma cell line MDA-MB-231, and immortalized mammary epithelial cell line MCF-10A. It revealed that both ACLY and p-ACLY were obviously higher in MCF-7 and MDA-MB-231 cells than in MCF-10A. Nevertheless, the difference is not significant in assessing the expression of ACLY and p-ACLY level between MCF-7 and MDA-MB-231 cells. Approximately 70% of BC tumors express ER; 43 thus, we chose to use the hormone-dependent, ER-positive human BC cell line MCF-7 for this study. 44 We determined the cellular functions of ACLY in MCF-7 cells. Our data clearly suggest that ACLY knockdown has anti-tumor activity in MCF-7 cells as evidenced by decreased cell viability and increased cell apoptosis. Abnormal ACLY signaling has been reported in many kinds of human cancers except BC. It was assumed that ACLY overexpression was necessary for tumor growth through the production of acetyl-CoA for lipogenesis; inhibition of ACLY effectively abolished tumor growth. Dysregulation of cellular metabolism is a hallmark of cancer cells. 45 Through suppression of ACLY in BC cells, we showed that elevated ACLY was essential for tumor growth. Because ACLY is a cross-link between glucose and lipid metabolism, it could play a potential role in molecular targeted therapy.
In conclusion, ACLY expression was up-regulated in BC, and ACLY knockdown inhibited tumor growth. We propose that inhibition of the ACLY-related pathway may represent a novel therapeutic strategy for controlling tumor growth in BC. Notably, many developed ACLY inhibitors offer new insight into potential clinical application.
Footnotes
Acknowledgements
The authors thank reviewers for critically reviewing the manuscript. They are also thankful to Jiangsu Key Laboratory of Translational Research and Therapy for Neuro-Psycho-Diseases, Institute of Neuroscience, Soochow University, Suzhou, P.R. China for supporting this research in part. They apologize to those whose works and related publications they have not been able to discuss and cite due to space limitations. The authors D.W. and L.Y. contributed equally to this work.
Compliance with ethical standards
Ethical approval was given by the medical ethics committee of the Second Affiliated Hospital of Soochow University National Clinical Medicine Ethics Committee with the following reference number: 20150909.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by grants from the Suzhou Science and Technology Bureau (KJXW2014 to Z.Y.). This project was subject to the Second Affiliated Hospital of Soochow University Preponderant Clinic Discipline Group Project Funding (XKQ2015008).