
Abstract
It is known that high-risk human papillomavirus infection is the main etiological factor in cervical carcinogenesis. However, human papillomavirus screening is not sufficient for early diagnosis. In this study, we aimed to identify potential biomarkers common to cervical carcinoma and human papillomavirus infection by proteomics for human papillomavirus–based early diagnosis and prognosis. To this end, we collected 76 cases of fresh cervical tissues and 116 cases of paraffin-embedded tissue slices, diagnosed as cervical squamous cell carcinoma, cervical intraepithelial neoplasia II–III, or normal cervix from ethnic Uighur and Han women. Human papillomavirus infection by eight oncogenic human papillomavirus types was detected in tissue DNA samples using a quantitative polymerase chain reaction. The protein profile of cervical specimens from human papillomavirus 16–positive squamous cell carcinoma and human papillomavirus–negative normal controls was analyzed by proteomics and bioinformatics. The expression of candidate proteins was further determined by quantitative reverse transcriptase-polymerase chain reaction and immunohistochemistry. We identified 67 proteins that were differentially expressed in human papillomavirus 16–positive squamous cell carcinoma compared to normal cervix. The quantitative reverse transcriptase-polymerase chain reaction analysis verified the upregulation of ASAH1, PCBP2, DDX5, MCM5, TAGLN2, hnRNPA1, ENO1, TYPH, CYC, and MCM4 in squamous cell carcinoma compared to normal cervix (p < 0.05). In addition, the transcription of PCBP2, MCM5, hnRNPA1, TYPH, and CYC was also significantly increased in cervical intraepithelial neoplasia II–III compared to normal cervix. Immunohistochemistry staining further confirmed the overexpression of PCBP2, hnRNPA1, ASAH1, and DDX5 in squamous cell carcinoma and cervical intraepithelial neoplasia II–III compared to normal controls (p < 0.05). Our data suggest that the expression of ASAH1, PCBP2, DDX5, and hnRNPA1, and possibly MCM4, MCM5, CYC, ENO1, and TYPH, is upregulated during cervical carcinogenesis and potentially associated with human papillomavirus infection. Further validation studies of the profile will contribute to establishing auxiliary diagnostic markers for human papillomavirus–based cancer prognosis.
Keywords
Introduction
Cervical cancer is one of the leading causes of cancer death among women in developing countries, with an estimated 450,000 new cases and 250,000 deaths each year.1,2 Invasive cancer accounts for more than 70% of patients diagnosed with cervical cancer, among which squamous cell carcinoma (SCC) is the most frequent type.2,3 Detection of cervical cancer at an early stage is crucial for effective treatment and associated with excellent survival rates, but women diagnosed with the advanced disease are often untreatable and have poor outcomes. 4 The Pap smear test is widely adopted as a cytological screening method for cervical dysplasia and precancerous lesions and may greatly reduce the incidence of cervical cancer in organized screening programs.5,6 Genital infection by human papillomaviruses (HPVs), in particular, by oncogenic high-risk HPV types (such as HPV16 and 18) is generally known as the main etiological factor in cervical carcinogenesis.7,8 Accordingly, HPV genotyping alone or with a Pap test is available as an alternative to cytology screening. However, most of the HPV infections are transient and harmless or eliminated by the host immunity. Even persistent infections have a relatively long latent period before inducing cancer, and thus, HPV testing may not improve cancer prognosis.9,10 The p16INK4a/Ki-67 dual staining cytology is introduced as a high-sensitive alternative for the screening of cervical intraepithelial neoplasia (CIN) that are high-risk HPV positive and Pap negative.11,12 However, since the expression of p16INK4a and Ki-67 is not dependent on HPV infection or progression to cervical carcinoma, the specificity of this dual staining is quite limited. 13 Thus, HPV-based screening or prognostication of cervical carcinoma may require the use of auxiliary diagnostic biomarkers that are highly associated with the infection by high-risk HPVs.
Proteins are executors of biological functions and important targets in cancer research in the post-genomic era. The advances in emerging proteomic techniques provide options for the screening of cancer-associated biomarkers and drug targets.14,15 The two-dimensional gel electrophoresis (2DE) is widely used in the profiling of cellular proteins, but it has low sensitivity. 16 With the development of a two-dimensional difference in gel electrophoresis (2D-DIGE), this technology is greatly improved in accuracy and sensitivity and successful in the identification of low-abundance proteins, such as potential tumor biomarkers. 17 The proteomics strategy based on isotopic labeling with isobaric tags for relative and absolute quantitation (iTRAQ) reagents currently allows for the analysis of up to eight different samples simultaneously and quantitative detection of very low–abundance proteins, such as HPV-derived proteins from cervical smears.18–20
Previous proteomics studies have identified a number of biomarkers for early diagnosis or prognostication of cervical carcinoma and its precursors. 21 Because high-risk HPV types are frequently detected in most of the cases of cervical carcinoma and precancerous lesions, these biomarkers for cervical carcinoma are possibly associated with the HPV infection. Nevertheless, protein profile changes common to cervical carcinoma and high-risk HPV infection have not been intensively studied.
In this study, the profile of differentially expressed proteins between HPV16-positive SCC and HPV-negative normal controls (NC) were investigated by iTRAQ-based proteomics followed by quantitative verification. The aim of this study was to further reveal the regulatory network of protein expression in cervical carcinoma that is associated with the HPV infection and identify auxiliary diagnostic markers for HPV-based cancer prognostication.
Materials and methods
Tissue specimens
The experimental protocol of this study was approved and monitored by the Ethics Committee of the Xinjiang Medical University, China. All procedures performed in this study were followed in accordance with the Helsinki Declaration of 1975, as revised in 2000. 5 The data were analyzed for all patients and healthy individuals anonymously throughout the study. Written informed consent was obtained from all individual participants included in the study. Patients with cervical SCC, CIN, and controls with normal cervix (NC) were enrolled in the study, according to the diagnostic criteria of the World Health Organization and the Chinese Medical Association. The median age of the patients was 43 (range: 26–63) years. In total, 76 fresh cervical lesions and normal cervix tissue from ethnic Han and Uighur women were collected by routine biopsies and surgical operations at the Department of Gynecology, the First Affiliated Hospital of Xinjiang Medical University. These specimens were pathologically classified as 36 cases of SCC, 15 cases of CIN II–III, and 25 cases of NC. Tissue specimens were snap frozen in liquid nitrogen after resection and stored in a −80°C freezer. For immunohistochemical (IHC) analysis, 116 cases of paraffin-embedded cervical specimens were obtained from the specimen bank in the Pathology Department of the First Affiliated Hospital of Xinjiang Medical University, China. These specimens were selected after a case review by two experienced pathologists and classified as 47 cases of SCC, 37 cases of CIN II–III, and 32 cases of NC.
The clinical staging of the patients was based on the guideline established by the International Federation of Gynecology and Obstetrics (FIGO) of 1994–1997, as revised in 1999 by Pecorelli et al. Tumor specimens were collected from cancer patients who underwent radical surgery for cervical SCC at clinical stages I–IIa. Precancerous lesions were collected from patients with CIN II–III as biopsies or tissue specimens from cervical conization. The tissue specimens classified as NC were obtained from patients without a history of cervical lesions or any form of cancer who underwent a hysterectomy for nonmalignant reasons during the same time period. Indications for hysterectomy were fibroids, prolapsed uterus, or adenomyosis, primarily a combination of fibroids with prolapse.
DNA extraction and HPV detection
Genomic DNA was extracted from fresh cervical specimens using a QIAamp DNA Mini Kit for tissue DNA (51306; Qiagen, Valencia, CA, USA) and quantified using a spectrophotometer (NanoDrop 2000; Thermo Scientific, Wilmington, DE, USA). The HPV infection was analyzed by a multi-fluorescent quantitative polymerase chain reaction (qPCR) assay using a Real Quality RQ-HPV HR kit (RQ-22-120A; AB Analitica s.r.l., Padova, Italy) with four probes for HPV genotyping of HPV16, HPV31, 18/45, and HPV33/52/58/67, respectively, on the ABI-7500 PCR system (Applied Biosystems, Carlsbad, CA, USA). The probe (primers) for human β-globin gene was used as an internal control for DNA quality.
Protein extraction
For proteomic analysis with 8-plex iTRAQ labeling, two groups of protein samples were prepared to separately analyze tissue specimens from ethnic Uighur or Han women. Each group contained four cases of HPV16-positive tumor specimens and four cases of HPV-negative normal cervix, respectively. A frozen tissue specimen was homogenized by grinding and transferred into a 1.5-mL tube that contained pre-chilled tricarboxylic acid (TCA)–acetone (500 mL acetone, 50 g TCA). After centrifugation (12,000 RCF, 4°C, 10 min) and discarding the supernatant, the precipitate was dissolved in a protein lysis buffer (8 M urea, 30 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 mM ethylenediaminetetraacetic acid (EDTA), and 10 mM dithiothreitol (DTT)). The protein sample (lysate) was then centrifuged and transferred into a new tube for removing the undissolved tissue residue. The DTT was added to the protein sample in a final concentration of 10 mM for reduction. After incubation for 45 min at 56°C, iodoacetamide buffer (55 mM IAA and 100 mM NH4HCO3) was added to the sample and incubated for 45 min for alkylation. The mixture was further mixed with pre-chilled 80% acetone at −20°C overnight and subsequently centrifuged for precipitation. The precipitate was dissolved in 300 µL dissociation buffer containing 50% triethylamine bicarbonate (TEAB) and 0.1% sodium dodecyl sulfate (SDS).
Tryptic digestion and iTRAQ reagent labeling
Each protein sample (100 µg) was digested with trypsin (protein:trypsin at 30:1) in a dissociation buffer (0.1% in TEAB; 37°C for 24 h). Eight peptide samples in each of the two groups (for ethnic Uighur or Han women) were labeled with 8-plex iTRAQ reagents (AB Sciex, Foster City, CA, USA) according to the manufacturer’s protocol (iTRAQ113, 114, 115, 116, 117, 118, 119, and 121). After removing the reaction solvent by speed vacuum, the labeled peptides were dissolved in 20 mM NH4FA (pH 10) for further analysis.
Peptide fractionation by strong cation exchange chromatography and reversed-phase chromatography
High-resolution strong cation exchange chromatography was used to remove the redundant iTRAQ reagents and any interfering substances that might affect the mass spectrometry analysis. Labeled peptides were loaded onto an SCX column (Luna SCX, 4.6 mm × 250 mm; Phenomenex, Torrance, CA, USA) and eluted by a stepwise linear elution program as follows: 0–10 min equilibration in Buffer A (25% acetonitrile (ACN), 20 mM KCl, and 10 mM KH2PO4, pH 3.0), 10–15 min fast elution in 0%–5% Buffer B (25% ACN, 1 M KCl, and 10 mM KH2PO4, pH 3.0), 15–50 min linear elution with 5%–30% Buffer B, and 50–55 min washing elution with 30%–80% Buffer B. For desalting and further fractionation, the peptide fractions were loaded onto an reversed-phase (RP) column (Luna C18, 4.6 mm inner diameter × 250 mm length; Phenomenex) and eluted by a step linear elution program as follows: 0–10 min equilibration in a 100% solution A (2% ACN and 20 mM NH4FA, pH 10), 10–15 min fast elution in 0%–12% solution B (80% ACN and 20 mM NH4FA, pH 10), 15–50 min linear elution with 12%–56% solution B, and 50–55 min washing elution with 56%–80% solution B. All of the procedures were performed using a Prominence HPLC system (Shimadzu, Kyoto, Japan) with a flow rate of 1.0 mL/min, and the peptides were monitored at 214 nm. The fractioned peptides were collected at a rate of one tube per minute during the linear elution period.
Peptide analysis by nano-liquid chromatography coupled with Q-Exactive mass spectrometry
The peptide fractions were loaded onto a nano RP column (5 µm Hypersil C18, 75 µm × 100 mm, Thermo Fisher Scientific Inc., Waltham, MA, USA) mounted in a Prominence nano HPLC system (Shimadzu). The peptides were eluted with an ACN gradient from 5% to 40% containing 0.1% formic acid for 65 min at 400 mL/min. The eluents were transferred to a Q-Exactive MS (Thermo Fisher Scientific Inc.), which was run in positive ion mode and a data-dependent manner with full MS scan from 350 to 6000 m/z, resolution at 70,000, tandem mass spectrometric (MS/MS) scan with a minimum signal threshold 17,500 and isolation at 2 Da. To evaluate the performance of the mass spectrometry on the iTRAQ-labelled samples, two MS/MS acquisition modes, higher collision energy dissociation (HCD) and collision-induced dissociation were employed. To optimize the MS/MS acquisition efficiency of HCD, the normalized collision energy was systemically examined from 25% to 70%.
Database search and quantitative data analysis
The raw MS/MS data were converted into Mascot Generic Format (MGF) format using Proteome Discoverer 1.3 (Thermo Fisher Scientific Inc.). The exported MGF files were searched by Mascot 2.3 (Matrix Science, Boston, MA, USA) against the Uniprot Human 2009–12 database with a precursor mass tolerance set at 15 ppm and production tolerance of 0.02 Da. An automatic decoy database search was performed. Carbamidomethylation of cysteines was set as a fixed modification (C) and oxidation of methionines (M), Gln to pyro-Glu (N-term Q), and 8-plex iTRAQ modifications of N-term, K, and Y were considered variable modifications. A maximum of one miscleavage was accepted. A protein with at least one unique peptide and a false discovery rate (FDR) < 0.01 qualified for further quantification analysis. The fold change in protein abundance was defined as the median ratio of all significantly matched spectra with tag signals. Based on analysis of Proteome Discoverer software, the coefficients of variation (CV) values of all quantified proteins and the quantitative results derived from duplicating injections were compared in parallel. The differential expression of all proteins was presented as the fold change in iTRAQ ratios. The upregulation of a protein was presented by the fold change of at least or more than 1.2 times and the downregulation by fold change of at least or less than 0.83 (1.0/1.2).
Bioinformatics analysis of MetaCore™ software
The differentially expressed proteins were further characterized by bioinformatic analysis using the software package MetaCore™ 6.18 (http://lsresearch.thomsonreuters.com/pages/solutions/1/metacore, Thomson Reuters) and its online database (https://portal.genego.com) to reveal the underlying pathways and protein–protein interaction networks and evaluate the candidate proteins as potential biomarkers.
RNA extraction and quantitative reverse transcription polymerase chain reaction analysis
The frozen tissue (~50 mg) was packed in aluminum foil and pulverized by grinding under liquid nitrogen. Total RNA was isolated from the powder by dissolving with TRIzol lysis buffer (Invitrogen, Carlsbad, CA, USA) followed by phenol/chloroform extraction and ethanol precipitation. The complementary DNA (cDNA) was synthesized from 1 µg of total messenger RNA (mRNA) by reverse transcription (RT) using a RevertAid Kit (MBI Fermentas, Burlington, ON, Canada) at 42°C for 60 min. For subsequent analysis, primer pairs specific to target mRNAs that were suitable for quantitative reverse transcription polymerase chain reaction (qRT-PCR) were designed and synthesized by the Takara Bio company (Takara Bio, Dalian, China). The cDNA (20 ng) was analyzed in a 25 µL mixture by qRT-PCR using an iQ5™ PCR system (Bio-Rad Laboratories Inc., Irvine, CA, USA) and SYBR Premix Ex Taq™ Kit (Ti RNase H Plus, Takara Bio, Tokyo, Japan). The qPCR reaction conditions were as follows: initial denaturation at 95°C for 30 s, followed by 40 cycles of denaturation at 95°C for 5 s and synthesis at 60°C for 30 s, and a final incubation at 4°C for 10 min. The expression level of target genes was quantified by the 2−ΔΔCt method using the internal software of the PCR system, setting β-actin as an internal control.
IHC analysis
Paraffin-embedded tissues were sectioned into 3 µm slices. The tissue slices were conventionally stained by the streptavidin peroxidase–conjugated (S-P) method using primary antibodies (Abcam, Bristol, England; Thermo Fisher Scientific Inc.) recognizing the target proteins and IHC Kits containing biotin-labeled secondary antibodies (Zhongshan Golden Bridge Biotechnology Co., Peking, China) in accordance with the manufacturer’s recommended procedures. The negative controls were stained with phosphate-buffered saline (PBS) instead of primary antibodies. All tissue slices were counterstained with hematoxylin.
For evaluation, stained tissue sections were scored under a light microscope by two experienced pathologists. The positive-stained area and intensity were scored using the following criteria. Positively stained area: score 0 for negative signal, 1 for 0%–30% positive, 2 for 31%–60%, and 3 for more than 60%. Positive intensity: score 0 for negative signal, 1 for weak signal intensity, 2 for moderate intensity, and 3 for high intensity. A consensus number between the two investigators was reached for each tissue slice. An overall score (0–6) was calculated by the addition of both scores to evaluate the expression level of a protein in three categories: an overall score of 0–2 as the loss or weak expression, 3–4 as moderate expression, and 5–6 as a strong expression of the protein.
Statistical analysis
Statistical analysis was performed with SPSS 17.0 for Windows (SPSS Inc., Chicago, IL, USA). All p values were two-sided, and the significance level was p < 0.05. The data derived from the qRT-PCR and IHC analysis were compared by one-way analysis of variance tests or paired samples t-tests. The specificity and sensitivity of potential biomarkers were determined by receiver operating characteristic (ROC) curve analysis.
Results
Identification of the protein expression profile associated with cervical carcinoma and HPV16 infection by nano-LC coupled with Q-Exactive MS
Among the 76 cases with fresh tissue specimens, HPV infection was detected in 52 cases by real-time PCR using genotyping probes for HPV16, 31, 18/45, and 33/52/58/67 (Table 1). Of these, 28 cases of SCC, 11 cases of CIN II–III, and 5 cases of NC were positive for HPV16 infection, respectively. For proteomics analysis by 8-plex iTRAQ labeling, eight cases of tissue specimens were selected as a group, including four cases of HPV16-positive SCC and four cases of HPV-negative NC. To exclude etiological factors possibly associated with the genetic background, two different groups of protein samples were prepared to separately analyze tissue specimens from the ethnic Uighur and Han women, respectively. The protein profile was investigated by nano-LC coupled with Q-Exactive MS.
HPV genotyping of cervical specimens.

HPV: human papillomavirus; NC: normal controls; CIN: cervical intraepithelial neoplasia; SCC: squamous cell carcinoma; n.d.: not detected.
To compare the overall proteomic profile distinguishing SCC from NC, we integrated peptide spectra that corresponded to four protein samples from SCC and NC in each of the two groups (for ethnic Uighur and Han women), respectively, using a functional module of the software Proteome Discoverer (version 1.3). Because the tissue specimens from SCC were almost identical in histopathologies, such as clinical stage (before stage IIa), differential state (low or moderate differentiation), and HPV infection, it was reasonable to consider either tumor specimens or NC in each group as biological repeats (data not shown). Subsequent analysis identified 67 differentially expressed proteins between SCC and NC, which was identical to the proteomics data for ethnic Uighur and Han women, setting a fold change of 1.2 times as the cut-off value for the differential expression. Of these, 33 proteins were upregulated (fold change > 1.2) and 34 proteins downregulated (fold change < 0.83 or 1.0/1.2) in SCC compared with NC, respectively (Table 2).
Differentially expressed proteins in HPV-positive SCC compared with HPV-negative normal controls.

HPV: human papillomavirus; SCC: squamous cell carcinoma; MW: molecular weight.
For a candidate protein, the fold change (iTRAQ ratio) of at least or more than 1.20 and at least or less than 0.83 (1/1.20) in cervical SCC compared with controls was considered and reported as up- and downregulation, respectively.
Samples from ethnic Han and Uighur women, respectively.
Bioinformatics analysis of the identified proteins and the prediction of potential biomarkers
The role of the identified proteins in disease development or carcinogenesis was further evaluated by bioinformatics analysis using MetaCore™ software (version 6.16) and its ontology database (http://www.genego.com). The gene ontology analysis showed that most of these proteins are localized in extracellular parts, intracellular organelles, or membrane-enclosed lumen (Figure 1) and involved in diverse cellular functions, such as glycolysis, gluconeogenesis, protein folding and maturation, or mitochondrial functions (Figure 2), and different biological processes, such as responses to injury repair, acute inflammatory reactions, or blood coagulation (Figure 3). The biomarker assessment analysis using the disease ontology database discovered that 16 of these proteins had been previously identified as potential biomarkers for cervical carcinoma. In accordance with our proteomics data, 11 of these proteins (Q03135, P07585, P08123, P20908, P02461, Q05707, Q9UBG3, P35555, P51884, P22105, and P21980) were downregulated and 5 proteins (P17844, Q9Y2Q3, P49736, P19338, and P12004) were upregulated in cervical carcinoma compared with NC. In addition, three proteins (Q03135, P19971, and P10909) were associated with SCC.
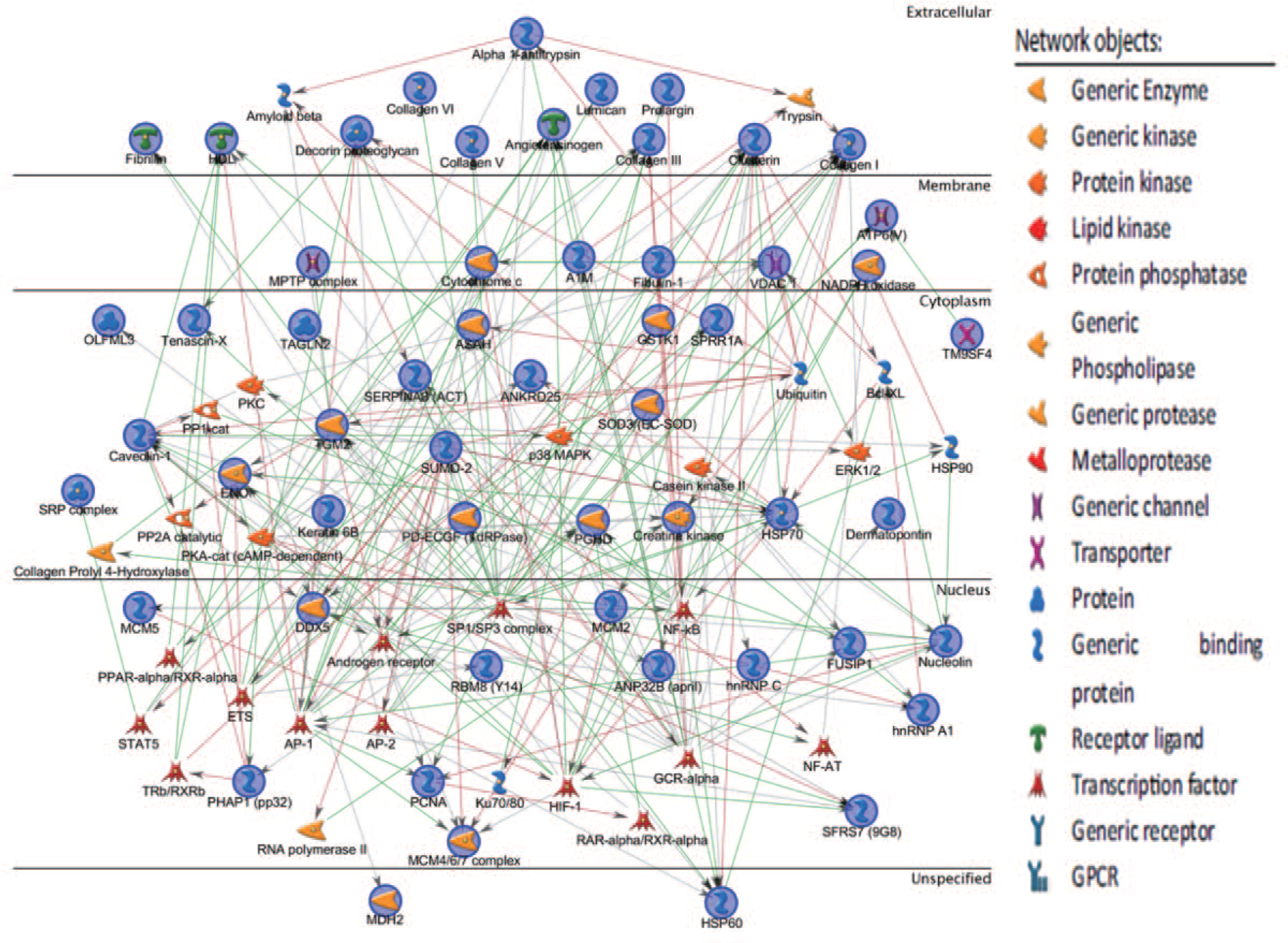
The network profiling of cellular signaling and gene expression for 67 candidate proteins by MetaCore™ software. The network includes all candidate proteins identified by proteomic analysis of tissue specimens from HPV-positive SCC and HPV-negative normal controls of the uterine cervix, as described in Table 2.

The distribution analysis of signaling pathways associated with 67 candidate proteins by MetaCore™ software. All candidate proteins described in Table 2 were included in the analysis.

Major biological processes associated with 67 candidate proteins as displayed by MetaCore™ software analysis. The description of all candidate proteins was shown in Table 2.
Verification of the profile of protein expression by quantitative RT-PCR and IHC
To verify the result of the proteomics analysis and identify novel biomarkers for cervical carcinoma, we selected 10 candidate proteins that were upregulated in the SCC: ASAH1, MCM5, CYC, MCM4, DDX5, PCBP2, TAGLN2, hnRNPA1, ENO1, and TYPH (see Table 2). Bioinformatics analysis and literature information indicated that the selected proteins might be associated with various cancers but had not been intensively studied in cervical carcinoma and its precursor lesions so far, with the exception of DDX5. The mRNA expression of these proteins was determined in 76 cases of cervical lesions and NC by qRT-PCR using primer pairs specific to mRNAs coding for the target proteins (Table 3). The results showed a significantly increased transcription of these genes in SCC compared with NC (p < 0.05), which was consistent with the proteomics data. In addition, significant upregulation was also found for PCBP2, MCM5, hnRNPA1, TYPH, CYC, and MCM4 in SCC as compared with CIN II–III, and for ASAH1, DDX5, and ENO1 in CIN II–III compared with NC (p < 0.05) (Table 4, Figure 4).
List of primers used in quantitative RT-PCR analysis.

RT-PCR: reverse transcription polymerase chain reaction.
Quantitative RT-PCR analysis of 10 proteins in cervical lesions.

HPV: human papillomavirus; NC: normal controls; CIN: cervical intraepithelial neoplasia; SCC: squamous cell carcinoma; ANOVA: analysis of variance; RT-PCR: reverse transcription polymerase chain reaction; mRNA: messenger RNA.
SCC, CIN, or NC refers to patients with cervical squamous cell carcinoma, cervical intraepithelial neoplasia, or normal cervix, respectively; SCC/CIN, SCC/NC, CIN/NC, comparative analysis of two groups indicated, with statistical significance at p < 0.05.

The transcription levels of 10 candidate genes in cervical lesions. (a–j) transcription levels of ASAH1, PCBP2, DDX5, MCM, TAGLN2, hnRNPA1, EON1, TYPH, CYC, and MCM4 in NC, CIN II-III, and SCC, as shown in Table 4, were displayed in bar graphs; *statistical significance by one-way ANOVA test at p < 0.05. The analysis included 10 candidate proteins identified by proteomics as described in Table 2.
We analyzed the expression of five candidate proteins, ASAH1, PCBP2, DDX5, hnRNPA1, and TAGLN2, by IHC to further verify the data of proteomics and qRT-PCR. The strong or moderate staining of PCBP2, hnRNPA1, and DDX5 was localized in the nucleus and ASAH1 in the cytoplasm of epithelial cells of cervical lesions, whereas TAGLN2 was exclusively found in vascular endothelial cells in the mesenchyme (Figure 5). The expression of PCBP2, hnRNPA1, DDX5, and ASAH1 was gradually increased in cervical specimens from NC to CIN II–III and SCC with significant differences (Table 5 and Figure 6) (p < 0.05). In the case of TAGLN2, no reliable statistical result was available because this protein was expressed in vascular endothelial cells instead of cervical epithelial cells in CIN II–III and controls and thus not comparable with SCC. Based on the data in Table 5, we evaluated the diagnostic potential of these proteins for cervical carcinoma by ROC curve analysis (Figure 7). The detection of ASAH1 in SCC, CIN II–III, and NC had a relatively high specificity (75.4%) but low sensitivity (57.4%), while PCBP2, DDX5, and hnRNPA1 were detected with relatively high sensitivity (83%–93.6%) and low specificity (42%–63.8%) (Table 6).

IHC staining pattern of five candidate proteins in cervical lesions. (a–c) typical expression patterns of ASAH1, PCPB2, DDX5, hnRNPA1, and TAGLN2 as weak expression in normal controls (a1–a5), moderate expression in CIN II–III (b1–b5), and strong expression in SCC (c1–c5), respectively. The description of candidate proteins was shown in Table 2.
Immunohistochemical detection of four proteins in cervical specimens.

HPV: human papillomavirus; NC: normal controls; CIN: cervical intraepithelial neoplasia; SCC: squamous cell carcinoma; ANOVA: analysis of variance.
SCC, CIN, or NC refers to patients with cervical squamous cell carcinoma, cervical intraepithelial neoplasia, or normal cervix, respectively; SCC/CIN, SCC/NC, CIN/NC, comparative analysis between two groups, with statistical significance at p < 0.05.
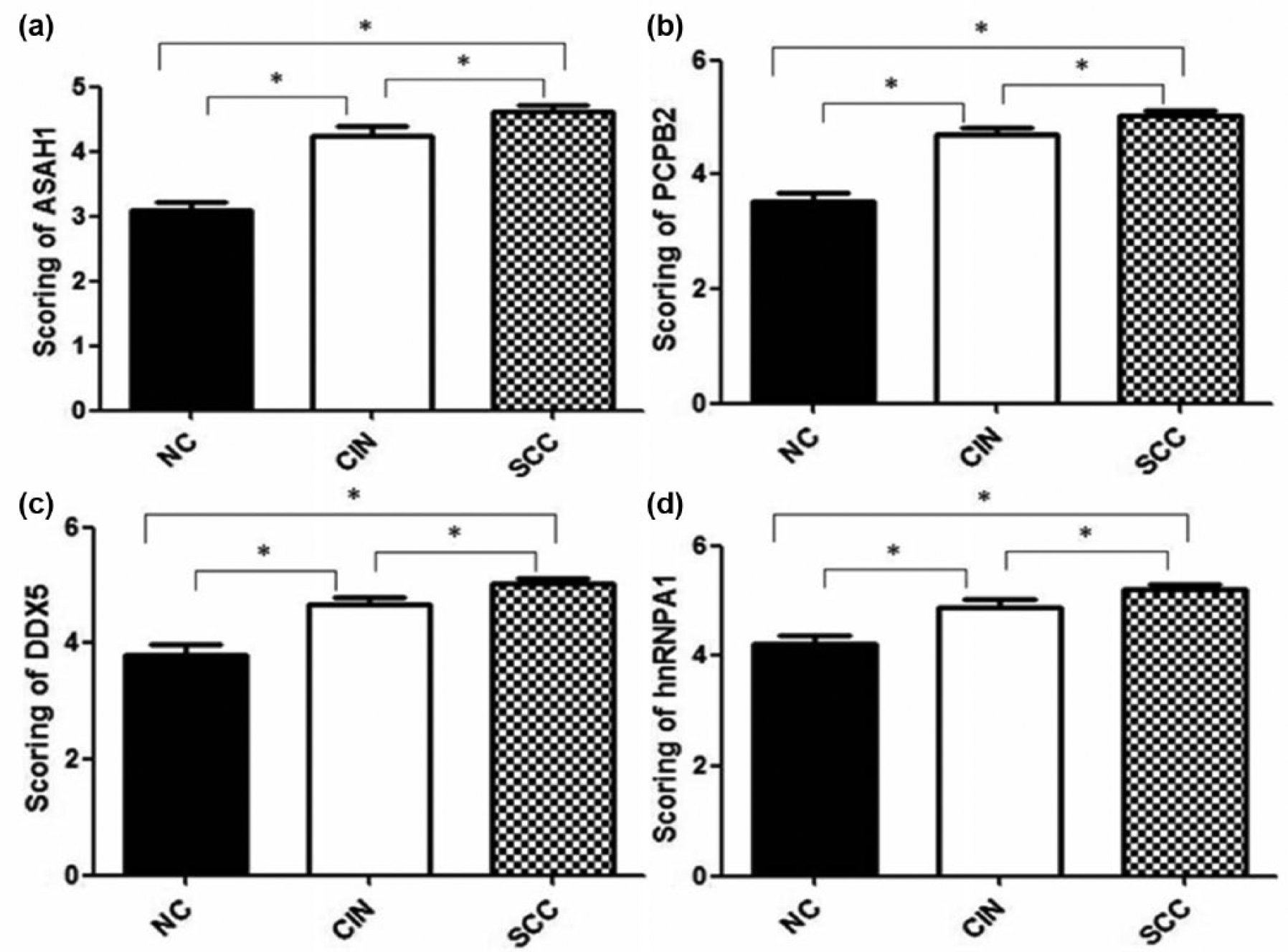
IHC-based analysis of protein expression for four candidate genes in cervical lesions. Protein expression levels of ASAH1, PCPB2, DDX5, and hnRNPA1 in NC, CIN II-III, and SCC, as shown in Table 5, were displayed in bar graphs (a–d); *statistical significance by one-way ANOVA test at p < 0.05. The analysis included four candidate proteins described in Table 2.

IHC-based ROC curves for four candidate proteins. The ROC curve analysis for ASAH1, PCPB2, DDX5, and hnRNPA1 (a–d), respectively, in the detection of NC, CIN II-III, and SCC by immunohistochemistry.
The sensitivity and specificity of four candidate proteins in the detection of cervical carcinoma.

Based on the analysis of HPV infection by detection of eight high-risk HPV types, all of the cases of SCC and most of the cases of CIN II–III were positive for HPV infection (100% and 73%), and most of the NC were HPV negative (80%) (see Table 1). Accordingly, we suggested that a possible correlation of protein expression with HPV infection may already be integrated into the results of the qRT-PCR and proteomics analysis. However, independent analyses are required to verify these results in the future.
Discussion
Proteomics strategies are frequently used to profile the protein expression of cervical carcinoma and its precursor lesions to identify tissue-based tumor biomarkers. Early studies showed the differential protein pattern between cervical SCC specimens and controls from the uterine cervix using 2DE technology and identified a number of proteins as potential biomarkers for SCC, such as HSP70, HSP27 (HSPB1), transgelin-2, eIF3B, NCF-2, ANXA6, TYK2, S100A9, and ZNF-217.22–25 The development of 2D-DIGE has improved the profiling sensitivity and accuracy for low-abundance proteins in the proteomics research of cancer. Recent studies based on 2D-DIGE have reported another set of proteins as possible tissue-based biomarkers for cervical SCC and precancerous lesions, such as S100A9, HSPB1, eEF1A1, FABP5, MnSOD, and cornulin.26,27 With the improved detection capacity for large numbers of low-abundance proteins by iTRAQ-based or shotgun proteomics, proteomics studies have revealed the complexity of the regulation network of protein expression during cervical carcinogenesis and identified novel biomarkers for SCC, including HSPB, G6PD, ALDH3A1, STAT1, KRT7, and KR17.28,29 In addition, redox proteomics identified the differential expression of ERp57, GST, TrxR2, and iNOS as a biomarker profile for HPV-positive invasive cervical carcinoma. 30 However, HPV genotyping was often not performed or considered in previous proteomics studies of cervical carcinoma. In our study, the protein expression pattern of HPV16-positive tumor specimens was compared with that of HPV-negative NC from the uterine cervix by 8-plex iTRAQ proteomics. We identified a profile of 67 differentially expressed proteins that were identical among two independent analyses of protein samples from the ethnic Uighur and Han women. Of these, 33 proteins were upregulated and 34 downregulated in SCC compared with controls. Subsequently, the profile was evaluated by bioinformatics analysis and verified by detection of 10 candidate proteins using quantitative RT-PCR and IHC.
As a result of bioinformatics evaluation, most of the identified proteins are functionally involved in glycolysis, gluconeogenesis, protein folding and maturation, mitochondrial functions, the cell cycle, or apoptosis. The biomarker assessment analysis predicted 16 proteins that are known biomarkers for cervical carcinoma or its precursors. Based on the proteomics data of this study, 5 of these proteins (GSTK1, MCM2, NUCL (NUNCL), DDX5, and PCNA) were upregulated and 11 proteins (PGS2, CAV1, CRNN, FBN1, COL1A2, COL5A1, COL3A1, COL14A1, LUM, TENX, and TGM2) downregulated in SCC compared with NC.
The upregulation of four predicted biomarkers, such as GSTK1, MCM2, NUCL, and PCNA, in cervical lesions or cells is reported by other studies. The expression of GSTK1 is downregulated in KB-8-5-11 cervical carcinoma cells in response to cisplatin treatment. 31 MCM2 is overexpressed in CIN and may serve as a predictive marker for cervical carcinoma. 32 NUCL may bind to the promoter of HPV18 in the S-phase and enhance the expression of viral-coding oncogenes in HPV18-positive cervical carcinoma cells. 33 PCNA is a coordinator of essential cellular functions for cell growth and maintenance. PCNA expression is upregulated in CIN and associated with high-risk HPV infections and has a predictive value for cervical carcinoma.34,35
The downregulation of 11 predicted biomarkers by bioinformatics was partially described by previous studies. PGS2/DCN is a newly identified soluble and matrix-derived tumor repressor that antagonizes signaling of various receptor tyrosine kinases (RTKs). 36 The overexpression of PGS2 may induce cell cycle arrest of SiHa cervical carcinoma cells. 37 CAV1 is an integral membrane protein of lipid rafts and a putative tumor suppressor gene that is downregulated in cervical lesions and cells transformed by HPV infection.38,39 CRNN is a marker of late epidermal differentiation and is downregulated in cervical lesions.25,30 FBN/LTPB is a calcium-binding protein of the extracellular matrix, and overexpression of FBN2 but not FBN1 has been reported in cervical adenocarcinoma. 40 LUM is a small leucine-rich proteoglycan expressed in the periphery of cancer nests of cervical SCC. 41 LUM expression is downregulated in prostate cancer, and its stromal expression is associated with reduced metastasis and prolonged survival of patients with pancreatic cancer.42,43 Contrary to the results of this study, the expression of TGM2, a multifunctional enzyme with transglutaminase crosslinking, G protein signaling, and kinase activities, is upregulated in cervical SCC and is associated with invasiveness. 44 Among the four tenascins (C, X, R, and W), glycoproteins that are located in the extracellular matrix, tenascin-C is known as an “oncofetal” protein controlled by many stimuli relevant to embryonal and fetal development, whereas tenascin-X (TENX) expression is barely affected by external factors. 45 Tenascin-C expression is markedly increased in invasive cervical carcinoma and oral SCC, but the role of TENX in tumorigenesis is largely unknown.46,47 Collagens are triple-helix proteins formed by the association of three polypeptide chains that are coded by different genes of the same collagen type. 48 Vertebrates have 28 different types of collagens that provide tissue-specific structure and function by assembling into a variety of supramolecular structures of the extracellular matrix. 49 Thus, the downregulation of several collagens, as shown in this study, indicates the dysfunction of extracellular matrices during tumor invasion and metastasis. COL5A2 is downregulated in cervical SCC and adenocarcinoma. 50 The coding genes for COL1A2, COL14A1, and COL15A1 are aberrantly methylated at the promoter region in a number of cancers, but not in cervical carcinoma.51–53 The COL3A1 expression is suppressed in nasopharyngeal carcinoma and upregulated by inhibition of miR-29 a/b. Its upregulation in renal cell carcinoma by ectopic expression of Let-7d micro RNA (miRNA) results in enhanced cell proliferation and migration.54,55 Based on the biomarker predictions from bioinformatics and previous findings, the differential expression of NUNCL, PCNA, and CAV1 is mostly associated with high-risk HPV infection in cervical SCC. Future studies are required to validate these markers. In particular, future studies need to investigate the association between downregulation of multiple proteins and promoter methylation of their coding genes to study possible novel mechanisms of HPV-induced carcinogenesis.
To verify the accuracy and specificity of our proteomics data, we determined the expression of 10 candidate proteins, namely, ASAH1, MCM5, CYC, MCM4, DDX5, PCBP2, TAGLN2, hnRNPA1, ENO1, and TYPH at both the transcriptional and protein expression levels. The transcripts coding for all of these proteins were significantly upregulated in cervical SCC as shown by qRT-PCR analysis. This was further confirmed by IHC analysis of four out of the five proteins, ASAH1, DDX5, PCBP2, and hnRNPA1.
TAGLN2 (TAGL2) is an actin-binding protein involved in cell morphology and transformation. 56 Concordant with our data from quantitative RT-PCR analysis, TAGLN2 expression is increased in cervical SCC and gastric adenocarcinoma but is downregulated in lung and breast carcinoma.22,57–59 Suppression of TAGLN2 inhibits proliferation and migration of cervical carcinoma cells. 60 Intriguingly, we found this protein to be located exclusively in the vascular endothelium by IHC and therefore could not verify the data of proteomics and RT-PCR analysis.
DDX5/p68 is an ATP-dependent RNA helicase and co-activator of transcription factors and thereby plays a role in RNA metabolism. 61 p68 is upregulated in SiHa cervical carcinoma cells in response to treatment with calcitriol (vitamin D3). 62 p68 overexpression may enhance β-catenin and androgen receptor signaling to promote cell proliferation in prostate and non-small-cell lung cancer cells.63,64 p68 acts as a co-activator of p53 and upregulates the expression of the oncogenic protein kinase, polo-like kinase 1 (PLK1) in the absence of functional p53, which in turn promotes mitosis in various cancers.65,66 However, not much is known about the regulation of DDX5 in cervical SCC.
ASAH1/AC is a lipid hydrolase that converts ceramides into sphingosine (SPH) and free fatty acids in different tissues. ASAH1 regulates the dynamic homeostasis of ceramides with sphingosine-1-phosphate (S1P), an anti-apoptotic signal that regulates cell maintenance and proliferation. 67 Accordingly, the elevation of ASAH1 results in an imbalance of this homeostasis between ceramide and S1P, which in turn promotes tumorigenesis. 68 ASAH1 is upregulated in epithelial ovarian cancer and breast cancer and is associated with a relatively good prognosis.69,70
Poly-cytosine-binding proteins (PCPBs) are RNA-binding proteins that regulate gene expression at multiple levels. 71 PCPBs have two main forms, the PCBP1 and PCBP2, with 82% homology in amino acid sequence. 72 PCBP1 is downregulated in various tumors; 73 in contrast, PCBP2 is upregulated in leukemia and glioma.74,75 In glioma and gastric carcinoma, PCBP2 interacts with tumor suppressors, such as FHL3, SIRT6, and miR-34a, and thus, its elevation supports tumor cell growth.76,77
HNRPA1 is a member of a family of RNA-binding proteins that are involved in a variety of functions, such as the regulation of transcription, mRNA metabolism, and translation. 78 hnRNPA1 may activate epithelial-to-mesenchymal transitions by interaction with the serine–arginine-rich splicing factor 1 (SRSF1) that regulates oncogenic alternative splicing. 79 Concordant with our results, increased hnRNPA1 expression is detected in cervical lesions, with the highest level in SCC cases that are positive for HPV infection. 80 In cervical epithelial cells immortalized with HPV16 E6 and E7, the hnRNPA1 expression is upregulated in response to hypoxia and glucose deprivation, typical features of the tumor microenvironment. 81
MCM proteins play important roles in cell cycle progression in mediating DNA replication initiation and elongation only once per cell cycle. MCM 4, 5, and 6 are differentially expressed in different types of cervical lesions, while MCM 4, 5, 6, and 10 are upregulated in SCC. In SCC, the overexpression of MCM 4, 6, and 10 is correlated with the advancement of tumor stages. 32 MCM detection in cervical smears has been suggested as an alternative to cytology screening of cervical lesions. 82
CYC is essential in mitochondrial electron transport and intrinsic type II apoptosis. This kind of apoptotic event involves changes in CYC phosphorylation, increased reactive oxygen species via increased mitochondrial membrane potentials, and oxidation of cardiolipin followed by CYC release from the mitochondria. 83 CYC expression is downregulated in response to radiation or treatment of cervical carcinoma cells with anti-cancer drugs.84,85 CYC expression is induced by ectopic expression of HPV16 E7 in HPV-negative cervical carcinoma cells, indicating that its expression is probably associated with HPV16 infection. 86 However, the upregulation of CYC in cervical lesions and SCC has not been reported previously.
ENO1 (enolase alpha) is a key glycolytic enzyme in the cytoplasm and on the surface of several types of cells, where it concentrates proteolytic activity by acting as a plasminogen receptor. 87 The expression of ENO1 is upregulated in gynecological cancers of the uterine cervix, ovary, and endometrium, and highest expression is frequently associated with brain metastases.88,89
TYPH (TP, ECGF1) is involved in thymidine metabolism and homeostasis by catalyzing thymidine dephosphorylation with broad substrate specificity. 90 TYPH is elevated in various tumors and plays a role in tumor progression and angiogenesis. 91 TYPH is highly expressed in microvessels of CIN and SCC and appears to be involved in angiogenesis in CIN. 92 TYPH activity is increased in invasive cervical SCC, with the highest level in lymph node metastases. 93
Based on these analyses, we demonstrated that the data of iTRAQ-based proteomics were not only supported by the qRT-PCR and IHC verification, and bioinformatic evaluation, but also highly consistent with earlier findings. However, due to the high HPV positivity of cervical lesions, namely, almost all specimens of SCC and most of the CIN II-III are positive for high-risk HPV genotypes, the analysis of gene expression for candidate proteins associated with HPV infection was very limited. Nevertheless, the proteomic identification of candidate proteins based on HPV genotyping was, to a very high extent, informative for understanding the mechanisms of HPV-driven cervical carcinogenesis.
As an alternative to HPV genotyping, the detection of viral oncogenic E6/E7 transcripts is considered as a potential marker of productive viral infection. 94 The high HPV E6/E7 mRNA expression is related to the severity of cervical lesions and a poor prognosis of cervical carcinoma, in contrast to the HPV DNA positivity that is not always associated with the histological grade of cervical lesions.95,96 Although both HPV DNA and E6/E7 mRNA can be detected at equally high rates in cervical carcinoma, the E6/E7 mRNA positivity is lower than HPV DNA in intraepithelial lesions.97,98 In addition, HPV E6/E7 mRNA assay may have higher specificity in the analysis of ThinPrep Pap Test samples, but lower sensitivity than HPV genotyping.99,100 These studies suggest that not all viral DNA copies are transcriptionally active in cervical lesions, and the E6/E7 mRNA assay may discriminate potentially progressive cervical lesions from transient dysplasias and provide useful information for early cervical cancer screenings.
Another limitation of this study was the analysis of HPV infection by only eight oncogenic HPV types (HPV16, 18, 45, 31, 33, 52, 58, and 67) using a method for clinical diagnosis. In accordance with the definition of International Agency for Research of Cancer (IARC) in 2009, HPV16, 18, 31, 33, 35, 45, 52, and 58 are carcinogenic to humans and most frequently found in cervical cancer, among which HPV16 is the most potent HPV type to cause human cancers, and HPV39, 51, 56, 59, and 68 are also carcinogenic and less constantly found in cervical cancer, while HPV26, 53, 66, 67, 70, 73, and 82 are considered “possibly carcinogenic” to humans in case of cervical cancer. 101 Therefore, the application of novel methods is required to detect other HPV genotypes and validate the result of this study in the future.
Conclusion
In this study, we show a profile of 67 proteins that are differentially expressed in HPV16-positive cervical SCC compared with HPV-negative NC. Since our proteomics data were confirmed by quantitative verification of nine candidate proteins and were mostly supported by previous findings of other studies, we suggest that the profiles, in particular, the upregulation of ASAH1, PCBP2, DDX5, and hnRNPA1, and possibly MCM4, MCM5, CYC, ENO1, and TYPH, are potential biomarkers for cervical SCC and high-risk HPV infection. However, the role of most of these proteins in HPV-induced carcinogenesis is largely unknown. Further validation of the profile, and the association between the altered expression of these proteins and HPV infection by other approaches, will contribute to establishing auxiliary diagnostic markers for HPV-based cancer prognosis.
Footnotes
Acknowledgements
S.Q. and W.T. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
The experimental protocol of this study was approved and monitored by the Ethics Committee of the Xinjiang Medical University, China. All procedures performed in this study were followed in accordance with the Helsinki Declaration of 1975, as revised in 2000. 5 The data were analyzed for all patients and healthy individuals anonymously throughout the study.
Funding
This study was supported by Natural Science Foundation of China (81360321). The funders had no role in the study design, data collection, analysis, decision to publish, or preparation of the manuscript.
Informed consent
Written informed consent was obtained from all individual participants included in the study.