
Abstract
Endometrial carcinoma is one of the most common gynecological malignancies, but the molecular events involved in the development and progression of endometrial carcinoma remain unclear. Dicer1 and cancer stem cells play important roles in cell motility and survival. This study investigated the role of the let-7 family and Dicer1 in the stemness of endometrial carcinoma cells. We profiled Dicer1 expression in clinical samples and explored its relationship with stem cell–associated markers and clinical parameters. We showed that Dicer1 dysfunction leads to the enrichment of tumor stemness features and tumor aggression both in vitro and in vivo. We also identified the mechanism related to this potential tumor-predisposing phenotype: loss of Dicer1 induced abnormal expression of the let-7 family, which comprises well-known tumor suppressors, thus regulating stemness in endometrial carcinoma cells.
Introduction
Endometrial carcinoma (EC) is one of the most common gynecological malignancies, ranking fourth among expected new cancer cases in women in the United States in 2016, with 10,470 estimated deaths. 1 However, the molecular events involved in the development and progression of EC remain unclear. To improve the outcomes of patients with EC, it is important to investigate the critical molecular pathways in the development of the disease and to identify novel therapeutic targets. 2
MicroRNAs (miRNAs) are ~22 nucleotide non-coding RNAs that regulate the translation and degradation of target messenger RNAs (mRNAs) and control approximately 30% of human genes. 3 Dicer1 is an endoribonuclease that plays a critical role in miRNA biogenesis. The two RNase III domains of Dicer1 form an intramolecular dimer that can cleave the pre-miRNA hairpin to generate mature miRNAs. These miRNAs can be combined into an miRNA-induced silencing complex (miRISC) and can interact with target mRNA. 4 As a single defect in the miRNA biogenesis pathway can potentially change the entire miRNA population, it is not surprising that interruptions of this pathway are associated with a variety of human diseases. 5 The role of Dicer1 in tumorigenesis has recently attracted increasing interest, and Dicer1 has been found as a key factor that determines a patient’s prognosis and the optimal therapeutic strategy in many types of cancer, such as ovarian cancer, lung cancer, or prostate adenocarcinoma.6–9 Recent studies have shown that colorectal cancer cells with impaired Dicer1 activity develop an enhanced stemness phenotype in association with a higher metastatic potential, which suggests an underlying connection between Dicer1 and cancer stemness. 10
The let-7 family of miRNAs are well-known tumor suppressors that repress cell proliferation pathways.11–13 In breast cancer, the let-7 family regulates the stem cell–like properties of multiple breast tumor initiating cells (BT-IC) by silencing H-RAS and HMGA2. 14 Recent studies have shown that Dicer1 hotspot mutations are present in ~2% of endometrial tumors. And Dicer1 mutations induce overexpression of great quantity of genes, of which many are targets of the let-7 family. 15
In our previous studies, we found that the expression of Dicer1 in EC was decreased. 16 We hypothesized that Dicer1 defects may contribute to human tumors by paving the way for generating cancer stem cells (CSCs) that are important for sustaining tumor growth. Here, we show for the first time that EC cells with impaired Dicer1 activity develop an enhanced stemness phenotype in association with decreased let-7 family miRNAs in EC.
Materials and methods
Tissue collection
Tissue samples for immunohistochemistry (IHC) were obtained from 83 patients with EC and 30 patients with normal endometrium who underwent surgical resection at the Department of Obstetrics and Gynecology, Shanghai General Hospital, between 2009 and 2012. The project was approved by the Institutional Review Board of Shanghai General Hospital, and informed consent was obtained from all patients before the study. The stages and histological grades of these tumors were established according to the criteria of the International Federation of Gynecology and Obstetrics. 17
IHC
For IHC staining evaluation, the staining intensity was scored as 0 (negative), 1 (weak), 2 (moderate), or 3 (strong). The staining percentage was scored as 0 (≤5%), 1 (6%–25%), 2 (26%–50%), 3 (51%–75%), or 4 (>75%). We obtained a composite histoscore by multiplying the values of the 2 parameters by a maximum of 12. Tissues with a final immunoreactivity score of ≥6 were considered positive for expression. Staining was scored independently by two pathologists without knowledge of the clinicopathological findings. The antibodies used for IHC were as follows: Dicer1 (ab11971; 1:5000; Abcam, Cambridge, UK), CD44 (3570; 1:50; CST), CD133 (MAB4399; 1:100; Merck Millipore, Berlin, Germany; CST, Washington, USA), Aldh1 (1:100; ab52492; Abcam), Ki67 (1:100; ab16667; Abcam), HMGA2 (1:100; ab97276; Abcam), and H-RAS (ab140377; 1:150; Abcam).
Cell culture
The EC cell lines RL-95, HEC-1B, and An3ca were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA), and Ishikawa was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). All cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Gibco, Life Technologies, Auckland, New Zealand) with 10% (v/v) fetal bovine serum (FBS) (Gibco, Carlsbad, CA, USA) at 37°C in a humidified atmosphere with 5% CO2.
Plasmid and transfection
An3ca cells were transfected with a Dicer1 full-length complementary DNA (cDNA) encoding plasmid, pDESTmycDICER (Addgene plasmid 19873, Addgene, www.addgene.org, provided by the laboratory of Dr Thomas Tuschl). The empty vector was used as a negative control. Ishikawa cells were transfected with short hairpin RNA (shRNA) against Dicer1 (shDicer1, Genechem, Shanghai, China) (sense: 5′-CAAGGAAATCAGCTAAATT-3′; antisense: 5′-AATTTAGCTGATTTCCTTGGC-3′) or shRNA against nontarget (NT; Genechem) by Lipofectamine™ 2000. After transfection, cells (IshikawashDicer1 and IshikawaNT cells) were selected in the presence of 0.5 µg/mL puromycin (Sigma-Aldrich, St. Louis, MO, USA) and maintained with 0.3 µg/mL. Overexpression and knockdown efficiency were confirmed by real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) and western blot.
hsa-let-7b mimics, hsa-let-7c mimics, and counterpart negative controls were synthesized by Shanghai GenePharma Co., Ltd, Shanghai, China. Ishikawa cells were transfected with the shDicer1 plasmid and with 50 nM hsa-let-7b (hsa-let-7c) mimics or negative control to generate IshikawashDicer1 + let-7b mi (IshikawashDicer1 + let-7c mi) or IshikawashDicer1 + let-7 con cells.
RNA isolation and qRT-PCR
Briefly, total cellular RNA was extracted using TRIzol® and TRIzol® LS Reagent (Life Technologies, New York, USA) following the supplier’s instructions. cDNA was generated using 1 mg total RNA and a QuantiTect Reverse Transcription Kit (Qiagen, Berlin, Germany). qRT-PCR of pre-miRNAs and the DUSP6 control was performed using SYBR Green (Bio-Rad Laboratories, Hercules, CA, USA) methods. The primers used are listed in Table 1. All primers and reagents for the pri-miRNA and mature miRNA analyses were purchased from Applied Biosystems (New York, USA) and performed according to the manufacturer’s instructions. Human β-actin or U6 snRNA served as an endogenous control for normalization. Other specific primers for qRT-PCR are also listed in Table 1.
Primers used for qRT-PCR analysis.
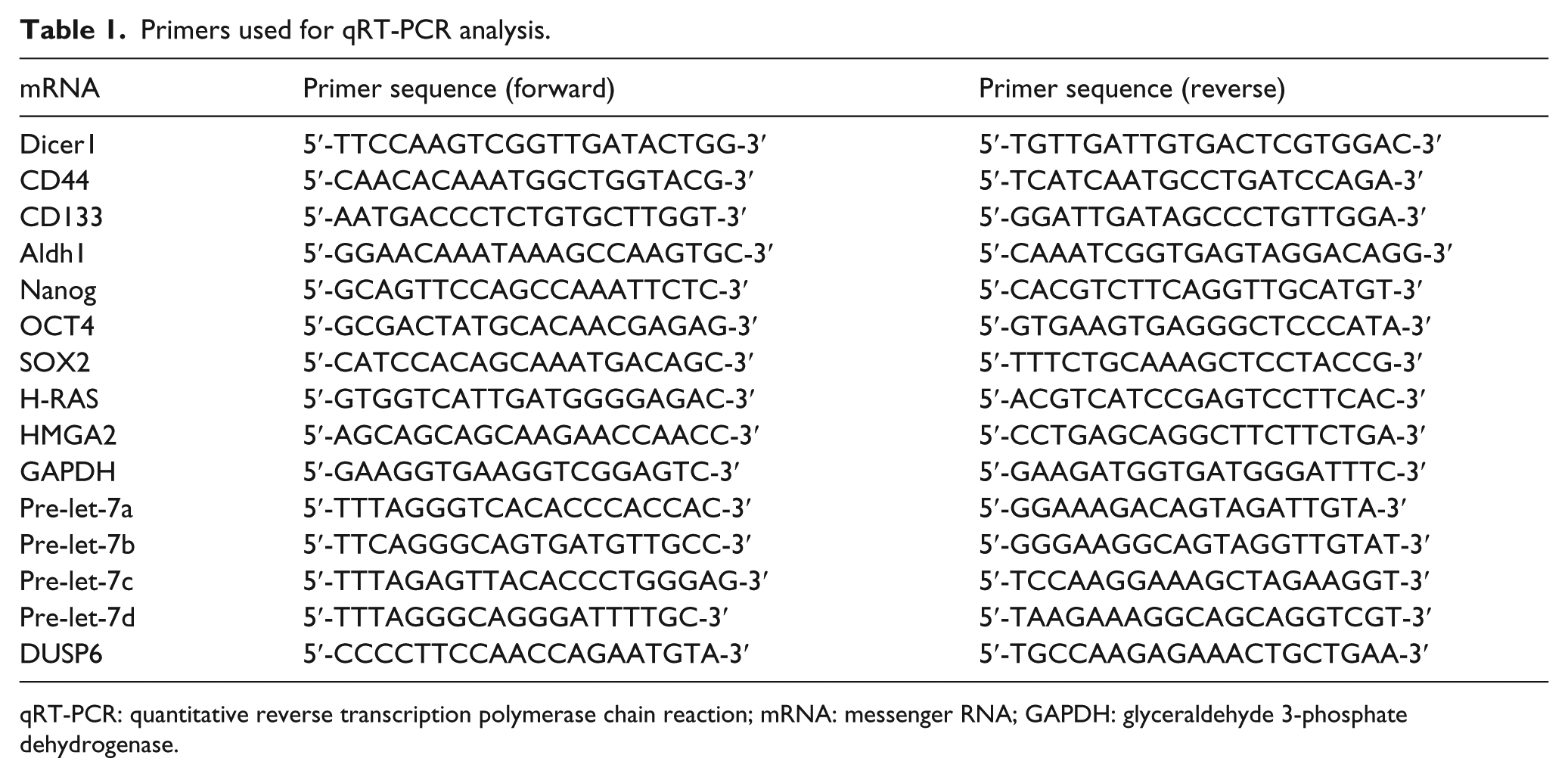
qRT-PCR: quantitative reverse transcription polymerase chain reaction; mRNA: messenger RNA; GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
Western blot analysis
Cells were lysed for total protein extraction using ProteoJET Mammalian Cell Lysis Reagent (MBI Fermentas, Ontario, Canada) including a protease inhibitor cocktail (Roche Diagnostics, Basel, Switzerland). Protein (80 µg) was loaded onto precast 4% stacking and 10% Tris-glycine gels, and separated by gel electrophoresis. Proteins in the gels were then transferred to polyvinylidene difluoride (PVDF) membranes. After transferring, membranes were blocked with 5% bovine serum albumin (BSA)/phosphate-buffered saline (PBS) for 3 h. The membranes were incubated with primary antibodies at 4°C overnight. The membranes were then incubated with peroxidase-linked secondary antibody (1:10000; Epitomics, Burlingame, CA, USA) for 2 h at room temperature. The blotted proteins were visualized using an enhanced chemiluminescence (ECL) kit (Beyotime, Beijing, China), and scanned and analyzed with TotalLab software. Primary antibodies included Dicer1 (ab11971; 1:2000; Abcam), Nanog (ab109250; 1:2000; Abcam), SOX2 (ab92494; 1:2000; Abcam), OCT4 (ab109183; 1:2000; Abcam), CD44 (3570; 1:200; CST), CD133 (MAB4399; 1:200; Merck Millipore), Aldh1(ab52492; 1:1000; Abcam), H-RAS (ab140377; 1:1000; Abcam), HMGA2 (1:1000; ab97276; Abcam), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 5632-1; 1:2000; Epitomics).
Cell immunofluorescence
Briefly, Ishikawa cells were seeded on the confocal laser dish at a density of 5 × 104 cells/mL and cultured for 24 h. The cultured cells were washed 3 times with PBS and fixed with 4% paraformaldehyde (Sigma-Aldrich) for 30 min. After blocking, the cells were incubated first with anti-CD44 (3570; 1:400; CST) or anti-Aldh1 (ab52492; 1:500; Abcam) antibody overnight at 4°C and then with Cy3-conjugated goat anti-rabbit IgG antibody (1:200; Abcam) and 5 mg/mL DAPI (4′,6-diamidino-2-phenylindole; Sigma-Aldrich) at room temperature for 30 min. Then, the cells were observed under a laser scanning confocal microscope (Leica, Heidelberg, Germany) with emission wavelengths of 518, 570, and 647 nm.
Cell proliferation assay
Cells were seeded in 96-well plates (3000 cells/well), and 10 µL CCK8 reagent (Obio Technology, Shanghai, China) was added to each well before incubation at 37°C for 3 h. The absorbance at 450 nm was measured using a SpectraMax 190 microplate reader (Bio-Rad Laboratories).
Colony formation assay
Cells were seeded in six-well plates (200 cells/well), and the plates were incubated at 37°C with 5% CO2 for 2°weeks to form colonies. The cell colonies were fixed with 4% methanol and stained with 5% crystal violet.
Mammosphere culture
Cells (1000 cells/mL) were cultured in suspension in serum-free DMEM-F12 medium supplemented with B27 (1:50, Invitrogen, New York, USA) and 20 ng/mL EGF (BD Biosciences, New York, USA). On the seventh day, the mammospheres were counted and measured under a low-power field inverted microscope.
Cell migration and invasion assays
Cell lines were suspended in serum-free DMEM-F12 medium and plated at a density of 1 × 105 cells/well (for the migration assay) or 2 × 105 cells/well (for the invasion assay) in 6.5 mm transwell chambers equipped with 8.0 µm pore polycarbonate membranes without or with Matrigel coating (BD Biosciences). Complete medium (600 µL) was added to the lower chamber. After incubation for 24 h (migration assay) or 48 h (invasion assay), cells were fixed in 4% paraformaldehyde and stained with crystal violet. Then, the cells that migrated to the basal side of the membrane were counted at 200× magnification.
Nude mouse tumor xenograft assay
Athymic female nude mice (BALB/c, 4- to 6-week-old, n = 5 per group) were obtained from Shanghai Life Science Institute (Slac Laboratory Animal Co., Ltd., Shanghai, China). IshikawashDicer1 or IshikawaNT cells were injected subcutaneously into the flank of each mouse at a density of 1 × 107 cells to establish a mouse model bearing EC cells. The growth of tumors was monitored throughout the experiment, and tumor size was measured with calipers every 7 days. The tumor volume was calculated as (RMax) × (RMin2) / 2. Five weeks after injection, mice were euthanized, tumors were removed carefully, and the weight and volume of the tumors were measured. All experimental protocols were approved by the Ethics Committee for Animal Experimentation of Shanghai Jiao Tong University.
Statistical analysis
All tests were carried out with SPSS 18.0 (Microsoft, Redmond, WA, USA) or Prism (GraphPad, San Diego, CA, USA). Each experiment was performed at least three times. When applicable, the data are shown as the mean ± standard deviation (SD). Two-tailed Student’s t-tests or Mann–Whitney U tests were used to compare data between two groups. Differences were considered statistically significant at p < 0.05.
Results
Dicer1 expression is low in EC specimens with high CSC marker expression compared with normal endometrium
We assessed Dicer1, CD133, and CD44 expression levels in 83 EC tissue samples and 30 normal endometrial tissue samples via IHC. Dicer1 protein was predominantly localized to the cytoplasm of endometrial epithelial cells, while CD44 and CD133 were localized to the membrane. We found strong Dicer1 staining in normal endometrium, whereas staining was moderate or weak in EC tissues. In contrast, the expression of CD44 and CD133 was increased in EC samples (Figure 1(a) and (b), *p < 0.05, **p < 0.01, ***p < 0.001).

Expression of Dicer1, CD44, and CD133 in EC tissues and cells. (a) Representative expression levels of Dicer1, CD44, and CD133 in normal endometrium and EC tissues. Original magnification 400×. (b) Immunohistochemistry scores (IS) of (a) (*p < 0.05; **p < 0.01; ***p < 0.001). (c) mRNA and protein expression of Dicer1 in EC cells as determined by qRT-PCR (**p < 0.01) and western blot, respectively. (d and e) Expression of Dicer1 was detected in IshikawaNT and IshikawashDicer1 cells or in An3caEV and An3caoeDicer1 cells by qRT-PCR and western blot (**p < 0.01).
We next explored the correlation of Dicer1 expression levels with the clinicopathological parameters of EC patients. As shown in Table 2, Dicer1 expression was lower in cases of deep myometrial invasion (p = 0.032), a high histological grade (p = 0.016), and lymph node metastasis (p = 0.004). In addition, Dicer1 expression was slightly decreased in high-grade EC specimens (p = 0.108). Moreover, Dicer1 expression was negatively correlated with CD44 expression, one of the CSC markers.
The relationship between Dicer1 expression and the clinicopathological features of EC.
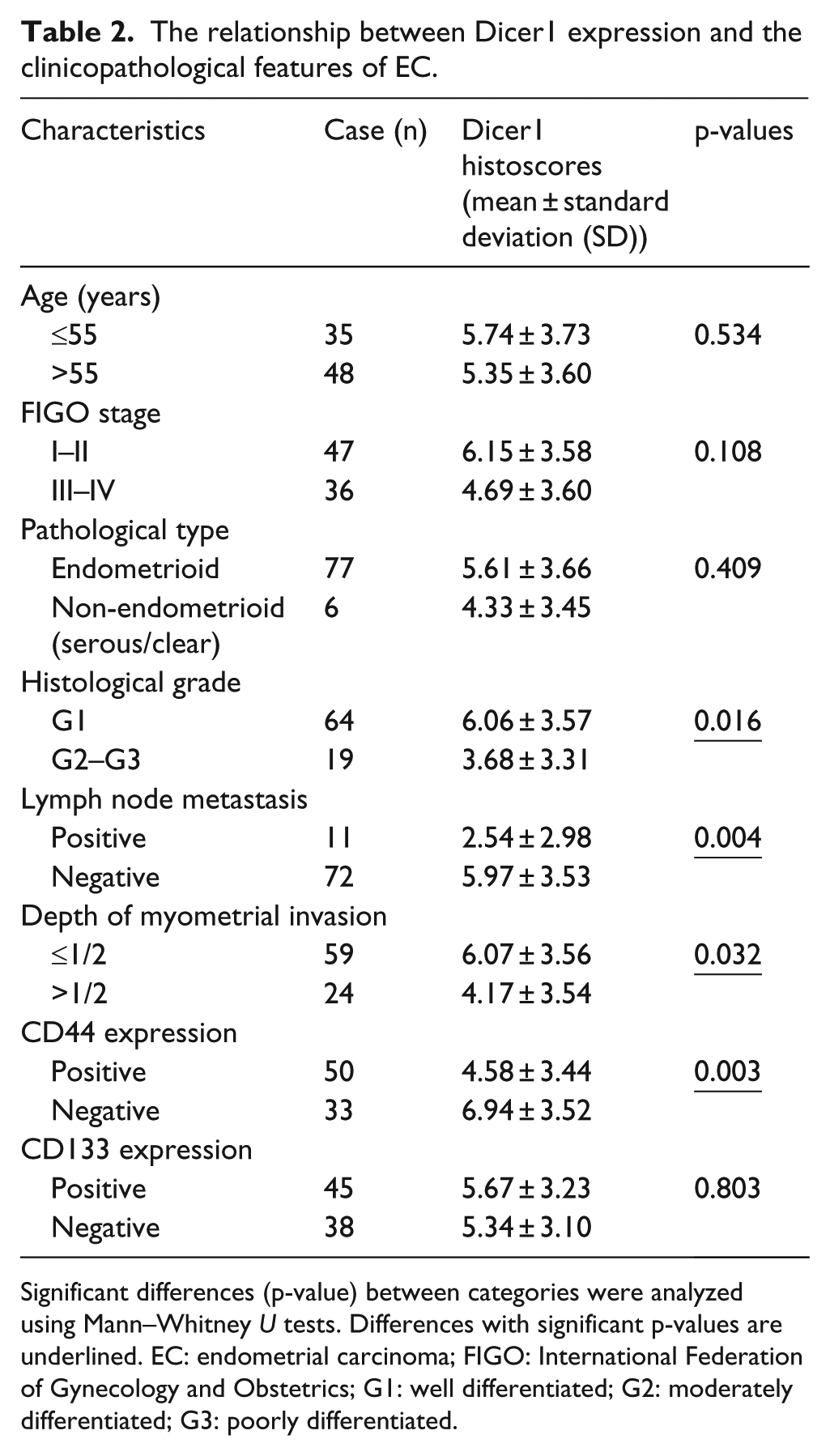
Significant differences (p-value) between categories were analyzed using Mann–Whitney U tests. Differences with significant p-values are underlined. EC: endometrial carcinoma; FIGO: International Federation of Gynecology and Obstetrics; G1: well differentiated; G2: moderately differentiated; G3: poorly differentiated.
These results suggested that Dicer1 expression, which was correlated with CSC marker expression, was associated with the development of EC and risk-associated clinical features of the disease.
Impaired Dicer1 function causes upregulation of stem cell markers in EC cells
To evaluate the effect of Dicer1 on the stemness of endometrial tumors, we first investigated Dicer1 expression across the EC cell lines by qRT-PCR and western blot (Figure 1(c)). Ishikawa (high expression) and An3ca (low expression) cell lines were chosen for further experiments based on their differential expression of Dicer1 (Figure 1(c), **p < 0.01).
To determine whether Dicer1 has an effect on the stemness of EC cells, we transfected Dicer1 expression plasmids into An3ca cells to generate Dicer1-overexpressing cells (An3caoeDicer1 cells) and transfected shRNA against Dicer1 plasmids into Ishikawa cells to generate Dicer1 knockdown cells (IshikawashDicer1 cells). As shown in Figure 1(d) and (e), the transfection efficiency of Dicer1 was confirmed by qRT-PCR after 48 h and by western blot after 72 h (**p < 0.01).
To further evaluate the effect of Dicer1 on EC stem cell markers expression, we detected the expression of the standard stem cell markers including CD44, CD133, Aldh1, OCT4, Nanog, and Sox2 using qRT–PCR and western blot. Compared with controls, knockdown of Dicer1 increased CD44, Aldh1, OCT, and Nanog expression in IshikawashDicer1 cells (Figure 2(a) and (c), *p < 0.05, **p < 0.01). In contrast, Dicer1 overexpression decreased the expression of CD44, ALDH1, Nanog, and OCT4 in An3caoeDicer1 cells (Figure 2(b) and (c), *p < 0.05, **p < 0.01). However, there was no change in CD133 and Sox2 between transfected cells and control cells. The differences in CD44 and Aldh1 between the transfected cell lines were also confirmed by immunofluorescence (Figure 2(d)). These results suggested that reduced Dicer1 expression may promote self-renewal of EC cells via their acquisition of stem-like features.
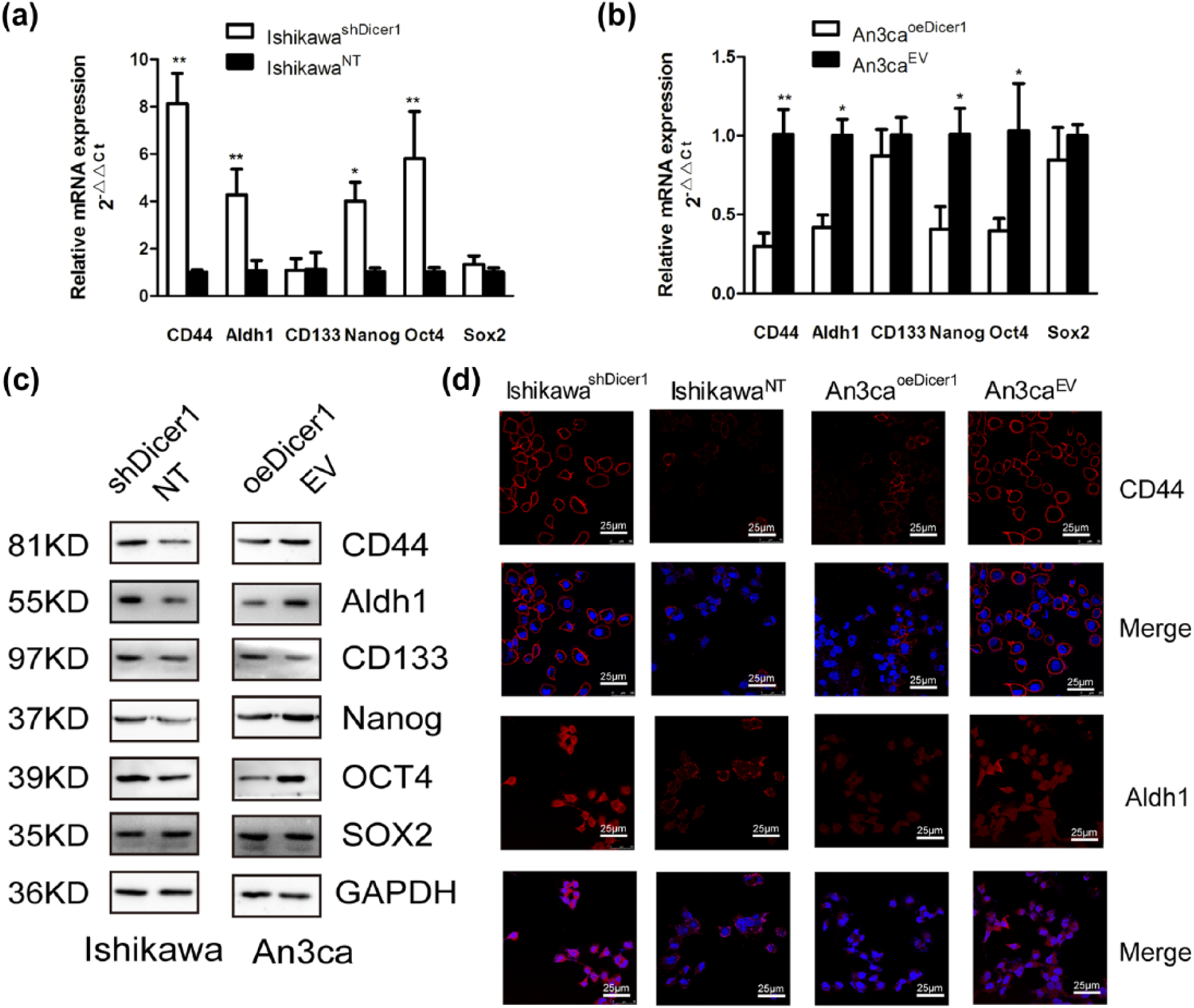
Impaired Dicer1 function causes upregulation of stem cell markers in EC cells. (a)–(c) mRNA and protein expression of CD44, Aldh1, CD133, Nanog, OCT4 and Sox2 was examined in IshikawaNT and IshikawashDicer1 cells or in An3caEV and An3caoeDicer1 cells by qRT-PCR and western blot (*p < 0.05; **p < 0.01). (d). Representative immunofluorescence images showing CD44 and Aldh1 expression in IshikawaNT and IshikawashDicer1 cells or in An3caEV and An3caoeDicer1 cells.
Dicer1 impairment promotes self-renewal and tumorigenicity of EC cells
As we demonstrated that Dicer1 was associated with the acquisition of stem-like features, we next explored whether this stem-like phenotype was associated with particular tumor biology properties. The most obvious approach was to assess its impact on tumor growth and metastasis.
We found that IshikawashDicer1 cells showed an increased viability as measured by CCK8 assays compared with IshikawaNT cells and that An3caoeDicer1 cells grew slower than control cells (Figure 3(a), *p < 0.05, **p < 0.01, ***p < 0.001). We also performed colony formation assays to further explore the role of Dicer1 in cell proliferation. We found more distinguished and frequent colony formation in IshikawashDicer1 cells than in IshikawaNT cells. In contrast, the overexpression of Dicer1 decreased the colony formation ability of An3ca cells (Figure 3(b), *p < 0.05, **p < 0.01). The mammosphere assay can identify stem-like properties, as it highlights the cells’ capacity for self-renewal and their ability to form a three-dimensional tumor-like sphere in vitro. We found that IshikawashDicer1 cells showed significantly increased spheroid formation capacity compared with control cells. Conversely, An3caoeDicer1 cells exhibited more spheroid formation capacity than control cells (Figure 3(c), *p < 0.05).

Dicer1 impairment promotes growth potential and invasion ability of EC cells. (a) The effect of Dicer1 on the cell growth of transfected cells was determined by CCK8 assays (*p < 0.05, **p < 0.01). (b) Plated colony-forming assays were performed to measure the cell proliferation of transfected cells. The colonies were counted and graphed (*p < 0.05). (c) Representative images of spheroid colonies are shown. Quantification and size of the spheroids formed by IshikawashDicer1 cells or An3caoeDicer1 cells compared to the control cells after 5 days (*p < 0.05). (d and e). Representative images of cell migration and invasion measured in transwell chambers. Quantification of migration and invasion at 24 and 48 h (*p < 0.05, **p < 0.01).
We next examined the effect of Dicer1 on EC cell migration and invasion. Transwell migration and invasion assays were performed to study the migration and invasion ability of EC cells. As shown in Figure 3(d) and (e), shRNA-mediated knockdown of Dicer1 in Ishikawa cells significantly increased cell migration and invasion compared with controls (Figure 3(d), **p < 0.01). Conversely, overexpression of Dicer1 in An3ca cells resulted in significantly decreased cell migration and invasion (Figure 3(e), *p < 0.05). These results indicated that Dicer1 was an important participant in EC cell invasion and metastasis.
Dicer1 acts as a tumor suppressor in a mouse tumor xenograft model
To further assess the role of Dicer1 in the progression of EC, we performed tumorigenicity assays in nude mice. Subcutaneous injections of IshikawaNT and IshikawashDicer1 cells in immunodeficient mice revealed that the tumors generated from the IshikawashDicer1 cells grew faster than those generated from the same amount of IshikawaNT cells (Figure 4(a) and (b), **p < 0.01). Then, the expression of CSC markers was investigated by IHC of tissues from tumor-bearing mice. Our results revealed significantly higher expression of CD44 and Aldh1 and higher expression of Ki67 in tumor tissues generated from the IshikawashDicer1 cells compared with the IshikawaNT cells (Figure 4(c)–(g), *p < 0.05).

EC stem cell markers were detected in tumor-bearing mice. (a and b) Five weeks after injection of the indicated cells, tumors were removed, and the tumor weights and volumes were determined (*p < 0.05; **p < 0.01; ***p < 0.001). (c) Representative Dicer1, CD44, Aldh1, and Ki67 immunohistochemical staining in tumor tissues generated from the IshikawashDicer1 cells or IshikawaNT cells. Original magnification 400×. (d)–(g) Quantification of (c) (*p < 0.05).
Let-7 family is involved in the regulation of Dicer1 to EC stemness
We showed that impaired Dicer1 function enhanced the stem cell features of EC cells. We next sought to identify a mechanism that might explain this phenotype. One possibility is that, within the spectrum of repressed synthesis of mature miRNAs in EC cells, miRNAs targeting key coding genes for stemness features would be markedly affected. The let-7 family comprises known tumor suppressors and participates in the regulation of stemness in breast cancer cells.14,18 In addition, previous studies have shown that, as a key enzyme in the biogenesis of miRNA, dysfunction of Dicer1 leads to low expression of the let-7 family. 19 In this regard, we next explored whether Dicer1 affects regulation of the let-7 family in EC.
We first detected let-7 family member expression in Dicer1 knockdown or overexpressing EC cells using RT-qPCR. The results showed that let-7 family member expression was low in Dicer1 knockdown cells and high in Dicer1-overexpressing cells as expected (Figure 5(a), *p < 0.05, **p < 0.01, ***p < 0.001). However, pri-miRNAs, the initial miRNA transcripts that are processed by DROSHA, were not significantly altered (Figure 5(a), not significant (NS)), and pre-miRNAs increased in Dicer1 knockdown cells and decreased in Dicer1-overexpressing cells (Figure 5(a), *p < 0.05, **p < 0.01), indicating that the effects on mature miRNAs were due to a defect in miRNA processing, not transcription. The level of mature let-7 family miRNA in the cells depended upon Dicer1 function.

Let-7 family is involved in the regulation of Dicer1 to EC stemness. (a) Let-7 family expression was changed in transfected EC cells. (b)–(d) Expression of CD44 and Aldh1 was detected in IshikawashDicer1 cells with let-7b (or let-7c) mimics or in control cells by qRT-PCR, western blot, and immunofluorescence (*p < 0.05; **p < 0.01).
To observe whether the let-7 family was involved in the regulation of EC cell stemness, we co-transfected let-7b mimics (or let-7c mimics) and its control with shDicer1 plasmids in Ishikawa cells. We then detected the expression of stem cell markers CD44 and Aldh1 using PCR, western blot, and immunofluorescence. The results showed that CD44 and Aldh1 expression was significantly reduced after re-expressing let-7b (let-7c) in the IshikawashDicer1 cells (Figure 5(b)–(d), *p < 0.05, **p < 0.01).
To observe the effect of let-7 on cell self-renewal and metastasis, we used CCK8 cell proliferation, colony formation, and sphere formation assays to observe cell self-renewal, and we used transwell migration and invasion experiments to observe cell metastasis. Our results showed that the self-renewal and metastasis capacity of the EC cells was significantly reduced for the let-7b (or let-7c) mimics (Figure 6(a)–(e), *p < 0.05, **p < 0.01).

Let-7 family inhibits growth potential and invasion ability of Dicer1 knockdown EC cells. (a)–(c). Effects of let-7b (or let-7c) on the cell growth of transfected cells were determined by CCK8 assays, plated colony-forming assays, and mammosphere culture assays (*p < 0.05; **p < 0.01). Representative images of plated colonies and spheroid colonies are shown. (d and e) Cell migration and invasion were significantly reduced in IshikawashDicer1 cells re-expressing let-7b (or let-7c) (**p < 0.01).
Dysfunction of Dicer1 changes the expression of let-7 target genes
As we showed that the re-expression of let-7 could reverse the upregulation of EC stem cell markers and the capacities for cell self-renewal and metastasis, which was mediated by dysfunction of Dicer1, we next investigated whether there was a change in let-7 target genes in Dicer1-transfected EC cells.
H-RAS and HMGA2 are target genes of let-7 and have been reported to participate in the regulation of cancer cell stemness in breast cancer. 14 Therefore, we used qRT-PCR and western blot to detect changes in H-RAS and HMGA2 in EC cells. As shown in Figure 7(a) and (b), knockdown of Dicer1 significantly increased H-RAS and HMGA2 protein expression in Ishikawa cells, and Dicer1 overexpression decreased H-RAS and HMGA2 protein expression in AN3CA cells (***p < 0.001). Neither treatment altered H-RAS and HMGA2 mRNA expression (Figure 7(c)). We also found that both H-RAS and HMGA2 expression levels were higher in the shDicer1 group compared to controls in vivo (Figure 7(d) and (e), *p < 0.05). These results suggested that let-7 family-related post-transcriptional regulation might affect the progression of EC. In addition, one manner in which Dicer1 may regulate EC stemness is via inhibition of the let-7 family, leading to upregulation of key target genes.

Dicer1 dysfunction changes the expression of let-7 target genes. (a)–(c) Expression levels of H-RAS and HMGA2 were examined in IshikawaNT and IshikawashDicer1 cells or in An3caEV and An3caoeDicer1 cells by qRT-PCR and western blot (***p < 0.001, NS). (d) Representative H-RAS and HMGA2 immunohistochemical staining in tumor tissues generated from the IshikawashDicer1 cells or IshikawaNT cells. Original magnification 400×. (e) Quantification of (d) (*p < 0.05).
Discussion
Dicer1 is an endoribonuclease that plays a critical role in miRNA biogenesis. It converts inactive hairpin-structured pre-miRNA into the active single-stranded form and then influences multiple cellular functions. 4 However, data regarding the correlation of altered Dicer1 expression and EC progression are scarce. 20
A current hypothesis in cancer cell biology suggests that tumor growth is sustained by a specific population of tumor cells, that is, CSCs, which exhibit features of adult stem cells, such as the ability to self-renew and differentiate. 21 To date, CSC markers that have been identified in EC include CD133, CD44, and Aldh1.22–25 Common CSC markers, such as SOX2, OCT4, and Nanog, have also been identified in other solid tumors. 26 In addition to expressing specific markers, CSCs are defined by their self-renewal capacity, their ability to generate new tumors and their increased capacity for migration and invasion. 21
A previous study reported that in colon cancer, Dicer1 knockout cells generated not only a cellular population that showed high levels of CD44 and enhanced tumor-initiating capability but also a specific subpopulation of CD44high/EpCAMlow cells endowed with metastasis-initiating activity. 10 These findings suggest that Dicer1 hypomorphic-mediated loss of miRNAs induces CSC properties associated with enhanced tumor dissemination.
Our IHC results indicated that, in EC tissues, Dicer1 expression was negatively correlated with CD44. Dicer1 knockdown EC cells exhibited significantly stronger expression of CD44, Aldh1, Nanog, and OCT4. We showed that knockdown of Dicer1 increased the proliferation, migration, invasion, and sphere-forming abilities of Ishikawa cells. Conversely, An3ca-overexpressing Dicer1 cells showed decreased ability of proliferation, migration, invasion and sphere-forming ability. We also discovered that Dicer1 knockdown significantly inhibited the growth of tumors in vivo upon conducting xenograft experiments.
Therefore, we first report that defects in Dicer1, one of the main enzymes involved in the generation of miRNAs, promote the stemness of EC cells.
Mounting evidence has shown that miRNAs are mutated or poorly expressed in human cancer, suggesting that miRNAs may act as tumor suppressors or oncogenes. 27 Several studies have demonstrated the role of specific miRNAs in regulating self-renewal, differentiation, epithelial mesenchymal transition (EMT) or metastasis of tumor cells.28,29 For example, increasing studies have reported Dicer1 and the let-7 family in gynecological neoplasms.15,30 The let-7 family is a well-known tumor suppressor, including 11 human let-7 family members, targets RAS and HMGA2,11,12 which encodes a DNA-binding protein implicated in mesenchymal cell differentiation and tumor formation. 31 It is well-known that the let-7 family regulates self-renewal and tumorigenicity of breast cancer cells.14,32 It was recently reported that let-7c inhibited estrogen-induced Wnt activity for the self-renewal of breast CSCs by decreasing ERα expression. 33 In EC, the expression of Dicer1 hotspot mutants promoted cell proliferation, and furthermore, targets of let-7 family miRNAs are enriched among the upregulated mutated genes, suggesting that loss of let-7 may affect downstream pathways. 15
Therefore, we investigated whether the let-7 family is involved in the regulation of Dicer1 in EC cell stemness. We co-transfected Ishikawa cells with shDicer1 plasmids and let-7b (or let-7c) mimics to observe the effect of the let-7 family on the regulation of EC cell stemness. We detected differences in the expression of stem cell markers CD44 and Aldh1 and cell self-renewal and metastasis capacity between let-7 mimic cells and control IshikawashDicer1 cells. The results showed that CD44 and Aldh1 expression as well as the self-renewal and metastasis capacity of EC cells were significantly reduced with the re-expression of let-7. Thus, the promotion of cancer cell stemness due to dysfunction of Dicer1 may be mediated by inhibiting let-7 family biogenesis.
To confirm this hypothesis, we also detected the expression of H-RAS and HMGA2, two key target genes of the let-7 family involved in the regulation of the stemness of breast cancer cell. Our results showed that Dicer1 knockdown significantly increased H-RAS and HMGA2 protein expression in Ishikawa cells, and Dicer1 overexpression significantly decreased H-RAS and HMGA2 protein expression in An3ca cells. However, there was no significant difference between the mRNA levels of HMGA2 and H-RAS, suggesting that let-7 family-associated post-transcriptional regulation may influence EC progression.
It was also recently reported that knockdown of Dicer dramatically increased cell resistance to camptothecin, which induced damage that required ataxia-telangiectasia mutated (ATM) gene to facilitate homologous recombination repair (HRR). 34 This phenotype was due to a prolonged G1/S transition via decreasing Dicer-dependent biogenesis of miRNA let-7, which increased p21Waf1/Cip1/p27Kip1 levels and resulted in decreased HRR efficiency. These results uncovered a novel function of Dicer in regulating the cell cycle through miRNA biogenesis, thus affecting the cellar response to DNA damage. Our data agree with the previous speculation that loss of let-7, a well-known tumor suppressor, might have a profound impact on downstream pathways. 35
A previous study showed that Dicer1 is also a target of let-7, while let-7 can potentially regulate Dicer expression. 36 Our results reveal that the Dicer1-let-7 axis has a marked influence on endometrial cancer stemness. In addition, let-7 biogenesis is Dicer1-dependent, as there was no significant difference in pri-let-7 levels, and the pre-let-7 levels changed conversely with mature let-7 members in Dicer1-transfected cells. In addition, the data also agree with a previous study by Chiappinelli et al., 19 which investigated the relationship between Dicer1 and interferon responses in EC cells. As there may be feedback regulation of Dicer1 and let-7, further studies are required to identify the detailed mechanism involved in the regulation of Dicer1 and let-7 on cancer cell stemness.
In summary, we report for the first time that Dicer1 dysfunction is associated with the stemness of EC cells. We show that specific miRNAs, such as those of the let-7 family, are extensively involved in regulating the self-renewal and tumorigenicity of malignant EC cell features. These findings suggest that Dicer1 combined with the let-7 family could potentially be utilized for EC therapy.
Footnotes
Acknowledgements
X.-J.W. and F.-Z.J. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The work was supported by the National Natural Science Foundation of China (NO.81302251, NO.81272885, NO.81472427, and NO.81672574) and the Natural Science Foundation of Shanghai (No. 15ZR1433400).