
Abstract
The 4-1BB is a surface glycoprotein that pertains to the tumor necrosis factor–receptor family. There is compelling evidence suggesting important roles for 4-1BB in the immune response, including cell activation and proliferation and also cytokine induction. Because of encouraging results of different agonistic monoclonal antibodies against 4-1BB in the treatment of cancer, infectious, and autoimmune diseases, 4-1BB has been suggested as an attractive target for immunotherapy. In this study, single chain variable fragment phage display libraries, Tomlinson I+J, were screened against specific synthetic oligopeptides (peptides I and II) designed from 4-1BB extracellular domain. Five rounds of panning led to selection of four 4-1BB specific single chain variable fragments (PI.12, PI.42, PII.16, and PII.29) which showed specific reaction to relevant peptides in phage enzyme-linked immunosorbent assay. The selected clones were successfully expressed in Escherichia coli Rosetta-gami 2, and their expression was confirmed by western blot analysis. Enzyme-linked immunosorbent assay experiments indicated that these antibodies were able to specifically recognize 4-1BB without any cross-reactivity with other antigens. Flow cytometry analysis demonstrated an acceptable specific binding of the single chain variable fragments to 4-1BB expressed on CCRF-CEM cells, while no binding was observed with an irrelevant antibody. Anti-4-1BB single chain variable fragments enhanced surface CD69 expression and interleukin-2 production in stimulated CCRF-CEM cells which confirmed the agonistic effect of the selected single chain variable fragments. The data from this study have provided a rationale for further experiments involving the biological functions of anti-4-1BB single chain variable fragments in future studies.
Introduction
The receptor 4-1BB (CD137 or tumor necrosis factor receptor superfamily member 9 (TNFRSF9)) was discovered more than two decades ago in 1989 after screening complementary DNA (cDNA) libraries on mouse activated CD4 and CD8 T-cell clones. 4-1BB is a 50 to 55-kDa surface glycoprotein concerned with progressive immunity that pertains to the tumor necrosis factor (TNF)-receptor family. 4-1BB genes encode type I cell surface glycoproteins comprised of an extracellular domain containing the amino-terminus of the protein, a transmembrane region, and an intracellular region.1,2
Expression of 4-1BB is inducible on the surface of activated T lymphocytes. Molecules that cross-link the T-cell receptor (TCR) or CD3 are sufficient to induce 4-1BB on both CD4 and CD8 cells. 4-1BB is stably upregulated when T cells are activated by a variety of agonists, such as anti-TCR monoclonal antibody (mAb), anti-CD3 mAb, concanavalin A, phytohaemagglutinin (PHA), interleukin (IL)-2, CD28, phorbolmyristate acetate (PMA), and ionomycin. However, 4-1BB is not detected (<3%) on resting T cells or T-cell lines. 4-1BB expression is not exclusive to activated T cells and has been detected on activated natural killer (NK) cells, B cells, and myeloid cells such as monocytes/macrophages, dendritic cells (DCs), eosinophils, mast cells, and neutrophils. In addition, 4-1BB expression has been reported on epithelial and hepatoma cells and in a constitutive manner on CD11c DCs and CD4 CD25 regulatory T cells.3,4
4-1BB binds to a high-affinity 4-1BB ligand (4-1BBL) that is the natural ligand for the 4-1BB receptor and, similar to OX40L, is constitutively expressed on a variety of antigen presenting cells (APCs), including DCs, B cells, and macrophages. It is induced hours or days after activation and can be regulated by lipopolysaccharide (LPS), immunoglobulin, or CD40 signals. 4-1BBL has the ability to costimulate T cells by triggering 4-1BB receptor. Moreover, it sends reverse signals to the APCs inducing proliferation, prolonging survival, and enhancing proinflammatory cytokines production. 4-1BBL was also detected on lymphomas, colon carcinomas, sarcomas, and melanomas. 5
CD28-B7 interaction plays a predominant role in the initial survival and proliferation of naïve T cells. However, within hours of activation, additional costimulatory molecules are upregulated and may serve to sustain, diversify, or differentiate the T-cell response. In recent years, 4-1BB and its ligand have been added to the growing list of costimulatory ligand–coreceptor pairs that can function independently of CD28 to activate T cells, as demonstrated by studies that engage 4-1BB with anti-4-1BB antibodies. Signaling through 4-1BB costimulates activation of CD4 and CD8 T cells in a CD28-independent fashion and provides survival signals via B-cell lymphoma 2 (Bcl-2) family members in T cells, particularly CD8 T cells. Signaling via 4-1BB by 4-1BBL, agonistic mAbs, soluble 4-1BBL, or cell lines expressing 4-1BBL regulates cell activation and proliferation of immune system during antigen-driven responses. Interaction of 4-1BB with 4-1BBL can induce T-cell expansion, cytokine induction, differentiation, and upregulation of anti-apoptotic genes as well as protect T cells from activation-induced cell death (AICD).6–8 4-1BB is mainly involved in regulating activated T lymphocyte functions and plays an important role in CD8 T-cell responses in a variety of systems, including viral infection, allograft rejection, autoimmune disease, and tumor immunity.9,10
In vivo studies have demonstrated the therapeutic potential of the 4-1BB/4-1BBL pathway in cancer and viral diseases. Activation of the 4-1BB/4-1BBL pathway in vivo using, for example, agonistic anti-4-1BB has led to increased survival, enhanced regression of tumor xenografts, or enhanced development of anti-tumor immunity in cancer bearing mice. 11 By adoptively transferring transgenic CD8 T cells into tumor-bearing RAG-2(−/−) mice, it was shown that the enhanced tumor rejection, delayed tumor progression, and prolonged survival can be mediated by CD8 T cells in the absence of CD4 T cells. Agonistic mAbs against CD137 have been shown to promote interferon gamma (IFN-γ) and TNF-α production, establish tumor rejection by CD8 T cells, and develop into antigen-specific cytotoxic T lymphocytes (CTLs). In tumor-bearing mice, enhancement of 4-1BB costimulation with IL-12 significantly amplifies both CTL and NK cell mediated immune responses and leads to the complete regression of established MCA26 colon cancer in a mouse model. The agonist human anti-4-1BB mAb is currently in Phases I and II clinical trials for the treatment of several solid tumors.12,13 In addition, it has been observed that administration of agonistic 4-1BB antibodies to lymphocytic choriomeningitis virus (LCMV)-infected or influenza-infected mice enhances T-cell expansion and survival in response to acute infection. These evidences suggest the use of agonistic 4-1BB antibodies in the setting of viral infections.14,15
In this study, we used phage display technology to isolate single chain fragment variable antibodies from a naïve phage library against two specific synthetic oligopeptides designed from 4-1BB extracellular domain. Five rounds of panning were performed, and selected single chain variable fragments (scFvs) were characterized using enzyme-linked immunosorbent assay (ELISA), DNA fingerprinting, western blotting, and sequencing. Moreover, the functionality of scFvs was examined by analysis of cell surface binding, CD69 expression, IL-2 messenger RNA (mRNA) expression, and IL-2 protein secretion.
Materials and methods
Materials and reagents
Human single fold scFv Tomlinson I+J libraries constructed in pIT2 phagemid were obtained from Cambridge, UK. In these libraries, scFv is cloned in the ampicillin resistant phagemid vector pIT2. M13KO7 was obtained from Source BioScience (Nottingham, UK) and used as a helper phage for biopanning. Peptides I and II were purchased from JPT (Berlin, Germany). Peptides were re-suspended in water (0.2 mg/mL), aliquoted, and stored at −20°C. Escherichia coli TG1 and E. coli Rosetta-gami2 were purchased from Novagen (Madison, WI, USA). CCRF-CEM cells were purchased from the National Cell Bank of Iran (Pasteur Institute of Iran, Tehran, Iran).
Anti-M13 and anti-C-myc mAbs conjugated to horseradish peroxidase (HRP) were purchased from Roche (Manheim, Germany). Anti-human CD3 mAbs were from Mabtech (Cincinnati, OH, USA). Human 4-1BB/TNFRSF9/CD137 antibody was purchased from R&D Systems (Minneapolis, MN, USA). Anti-CD69 mAb conjugated to phycoerythrin (PE) was purchased from BioLegend (San Diego, CA, USA).
Taq DNA polymerase, deoxynucleotides (dNTPs), and DNA ladders were from Takara (Dalian, China). DNA restriction enzyme was from Thermo Scientific (Waltham, MA, USA). DAB (3, 3′-diaminobenzidine), bovine serum albumin (BSA), ampicillin, kanamycin sulfate, isopropyl b-
Library amplification
A volume of 500 µL of phage display libraries, Tomlinson I (library size: 1.47 × 108), and Tomlinson J (library size: 1.37 × 108) were incubated in 200 mL of 2XYT containing 120 µg/mL ampicillin at 37°C and 250 r/min until the OD600 = 0.4 − 0.5. Then, about 5 × 1011 cfu (colony-forming units) per milliliter of helper phage M13K07 was added and incubated for 1 h at 37°C. After that, bacteria were collected by centrifuging for 10 min at 4000 r/min, re-suspended in 100 mL of the fresh 2XYT with 120 µg/mL ampicillin and 50 µg/mL kanamycin, and incubated overnight at 37°C. Bacterial cells were removed by centrifugation at 4000 r/min for 20 min at 4°C, and subsequently phages were precipitated from the supernatant with polyethylene glycol (PEG)-NaCl.16,17
Screening of Tomlinson’s library for 4-1BB specific scFv
In order to select specific anti-4-1BB scFvs, biopanning technique was performed. Immunotubes were coated with two specific peptides designed from 4-1BB extracellular domain, peptide I: 14 amino acids (TKKGCKDCCFGTFN) and peptide II: 14 amino acids (GTKERDVVCGPSPA), as well as BSA as a negative control at a concentration of 10 µg/mL in phosphate-buffered saline (PBS) and incubated overnight at 4°C. The immunotubes were washed twice with PBS and blocked with 5% MPBS (5% skim milk in PBS) for 90 min at 37°C. Afterward, blocking solution was removed and the prepared phages from the library (1012 pfu (plaque forming units)/mL) were mixed with 5% MPBS for total volume of 4 mL and added to BSA-coated tubes for 1 h at 37°C and then transferred to the 4-1BB peptides-coated tubes and incubated for 1 h at 37°C. The immunotubes were washed 10 times with PBS containing 0.5% Tween-20, five times with PBS, and finally with distilled water to remove the unbound phages. Then, PI and PII specific phages were eluted with 100 mM triethylamine (TEA) and neutralized with Tris–HCl. Afterward, for amplification of the phage particles, competent E. coli TG1 cells (OD600 = 0.5) were infected with the eluted phages and M13KO7 helper phage. Five consecutive rounds of panning were performed to isolate specific antibodies against the target peptides. 18
Polyclonal scFv-phage ELISA
The binding activity of the purified scFv products was determined by ELISA. After the fifth round of panning, obtained phages were grown in 200 mL lysogeny broth (LB) containing 120 µg/mL ampicillin. Then, phages were produced by addition of M13K07 helper phages and 50 µg/mL kanamycin. High affinity 96-well ELISA plates were coated with 10 µg/mL of specific peptides (PI and PII), and c-Met (hepatocyte growth factor receptor) peptide and BSA protein were used as negative controls. The plates were blocked at 37°C for 90 min with 250 mL of 3% w/v BSA in PBS buffer. After blocking, phage-rescued supernatants (1012 cfu/mL) were added to the plate and incubated at 37°C for 60 min. The plates were washed four times with PBS containing 0.05% Tween-20 (pH 7.2). After the removal of unbound phages, 100 µL of the HRP-conjugated murine anti-M13 (1:5000 v/v) was added to each well and incubated at 37°C for 60 min. Following incubation, the plates were washed thrice with PBS containing 0.05% Tween-20 and one time with PBS, and then, 100 µL of tetramethylbenzidine (TMB) substrate was added to each well and the plates were incubated in the absence of light at room temperature (RT). The enzyme–substrate reaction was stopped by adding 50 mL of 1 M sulfuric acid to each well. ODs were read at 450 nm using an ELISA reader (BP-800; Biohit Inc., Neptune, NJ, USA).19,20
Screening of individual scFv-phage clones by ELISA
Two hundred phage clones (100 clones for PI and 100 clones for PII) were randomly selected from output phages and tested for 4-1BB binding by monoclonal phage ELISA as the protocol for polyclonal phage ELISA. Each of phage clones were grown in LB containing 120 µg/mL ampicillin and 100 µg/mL kanamycin. Then, phages were produced by addition of M13K07 helper phages. High affinity 96-well ELISA plates were coated with 10 µg/mL of specific peptides (PI and PII). Background binding of the phage clones to the ELISA plate was defined by using antigen-free wells.21,22
Production of soluble 4-1BB scFvs
Individual scFv clones that are shown to be positive in monoclonal phage ELISA were used to obtain soluble scFvs. The log-phase E. coli Rosetta-gami 2 was infected with the strongest positive phage particles and grown at 37°C in 2XYT medium supplemented with 120 µg/mL ampicillin until an OD600 of 0.9 was achieved. The scFv expression was induced with 1 mM IPTG and incubated at 30°C for 18 h. The bacteria were subsequently harvested by centrifugation at 3700 r/min for 15 min at 4°C and re-suspended in PBS with 1 mM PMSF. Anti-4-1BB antibodies existing in the periplasm were extracted by standard osmotic shock procedure. For this, the pellets were suspended in ice-cold Tris–EDTA–sucrose (TES) buffer (hypertonic buffer) and incubated on ice for 1 h. Then, four times diluted TES buffer (hypotonic buffer) was added and incubated on ice with shaking for 2 h. After centrifugation at 10,000 r/min for 30 min at 4°C, the supernatant containing the periplasmic protein fractions were collected and analyzed for the presence of soluble antibody by ELISA. An HRP-conjugated anti-C-myc antibody was used as the secondary antibody (1:2000 dilution).21,23
Sequence analysis of the positive clones
Polymerase chain reaction (PCR) was carried out to check the selected ELISA positive clones for the presence of full-length VH and VL inserts. The scFvs from the Tomlinson library were amplified with primer pairs LMB3 (5′ CAGGAAACAGCTATGAC 3′) and pHEN (5′ CTATGCGGCCCCATTCA 3′). PCR products were examined by electrophoresis on 1.5% agarose gel containing Green viewer in TAE (Tris-acetate-EDTA) buffer. Genetic diversity of the positive clones was determined by fingerprinting with BstNI restriction enzyme. Moreover, DNA bands were purified with the QIAquick Gel Extraction Kit (Qiagen GmbH, Hilden, Germany) and sequenced by Bioneer Corporation (Daejeon, South Korea).24,25
Analysis of selected scFv clones by western blotting
The soluble scFv antibodies from the periplasmic extract of E. coli Rosetta-gami 2 were quantified by Bradford assay and equal amounts of protein were separated on a 10% discontinuous sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). Following PAGE, the separated proteins were transferred onto a nitrocellulose membrane. The membrane was subsequently blocked overnight with 3% BSA at 4°C and then incubated in 2000-fold-diluted HRP-conjugated anti-C-myc for 2 h at 37°C. After washing three times with 0.05% PBST and PBS, the bound antibody was detected by staining with DAB.25,26
Specificity
The ELISA test was performed with a variety of antigens to assess cross-reactivity of positive soluble scFvs. 4-1BB, insulin-like growth factor 1 receptor (IGF-1R), hepatocyte growth factor receptor (c-Met), receptor tyrosine kinase-like orphan receptor 1 (ROR1), programmed cell death protein 1 (PD1), skimmed milk, and BSA were coated onto 96-well ELISA plates at 10 µg/mL. Wells were blocked and then incubated with selected scFvs. This test was performed similar to the previous ELISA test with the HRP-conjugated anti-C-myc antibody.27,28
Cell binding analysis by flow cytometry
Cell surface binding of the scFvs was measured by a Partec PASIII (Partec GmbH, Münster, Germany) flow cytometer. The CCRF-CEM cells were cultured in RPMI 1640 medium supplemented with 15% fetal bovine serum (FBS), 100 µg/mL streptomycin, and 100 U/mL penicillin. The cells were stimulated with anti-CD3 antibody plus IL-2 for 48 h at 37°C in a humidified CO2 incubator. Then, CCRF-CEM cells were washed twice with PBS−5% FBS. Thereafter, 5 × 105 CCRF-CEM cells (cell viability was 95% as determined by trypan blue exclusion) were re-suspended and incubated in buffer alone or buffer with selected phage-scFv antibodies (1012 cfu/mL) for 45 min at 4°C. After two washes with buffer, cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-M-13 antibody for 45 min at 4°C. Stained cells were analyzed using FlowJo 7.6.1 software (Tree Star, Inc., Ashland, OR, USA). In a parallel set of experiment, the cells were also stained with a commercial goat anti-4-1BB mAb and FITC-conjugated anti-goat antibody as a positive control and with an anti-c-Met scFv as an isotype control.29,30
Determination of IL-2 mRNA by real-time PCR
The mRNA levels of IL-2 were assessed using real-time PCR. Total RNA was extracted from cells treated with PI.12, PI.42, PII.16, and PII.29 scFvs, and an unrelated scFv (named ES1) using the RNeasy Mini Kit (Qiagen). Total RNA (1 mg) from cell lines was used to synthesize cDNA using cDNA synthesis kit (Roche, Gipf-Oberfrick, Switzerland), according to the manufacturer’s instructions. The expression levels of mRNA were determined using Two-Step Quantitech SYBR Green RT PCR kit (Takara) in the Corbett Rotor-Gene 6000 (Corbett Life Science, Sydney, Australia). Designated primers for IL-2 were 5′-ATGTACACCATGCAACTCCTGTCT-3′ and 5′-GTCAGTGTTGAGATGATGCTTTGA-3′. Hypoxanthine-guanine phosphoribosyltransferase (HPRT) was employed as the housekeeping control to analyze mRNA expression. Changes in IL-2 expression after treatment were calculated using the 2−ΔΔCtmethod.31,32
IL-2 production assay
To measure cytokine production, CCRF-CEM cells were stimulated with anti-CD3 antibody and IL-2 as described in the previous section. After 48 h, the cells were treated in the presence or absence of the selected scFvs and IL-2 secretion was evaluated in supernatants of treated and control CCRF-CEM cells using standard sandwich ELISA kits (R&D Systems).31,32
Assessment of CD69 expression
To demonstrate the activation state of CCRF-CEM cells, the expression of CD69 was measured. Briefly, after a 24-h treatment with PI.12, PI.42, PII.16, PII.29, and ES1 scFvs, 106 cells were washed twice with washing buffer (PBS 0.15 M, 0.5% BSA) and stained with anti-CD69 PE mAb. After 30 min of incubation at 4°C in dark, the cells were washed and analyzed by flow cytometry. 31
Statistical analysis
One-way analysis of variance (ANOVA) and t tests were used for statistical analysis of the data and calculation of p value. Results are expressed as mean ± standard deviation (SD).
Results
Enrichment of 4-1BB specific binders
Biopanning against the peptides I and II was performed using the Tomlinson I+J libraries to select 4-1BB-specific scFvs. Five rounds of panning were carried out on 4-1BB antigen. The stringency of the selection was increased by progressively increasing the Tween-20 concentration (0.5%–8%) in washing buffers. Enrichment of the 4-1BB-specific scFvs was tested by measuring the ratio between the input number of phage particles at the beginning of each experiment and the number of phage particles recovered at the end. After the first round of selection, the number of phage particles recovered was 104 cfu/mL for PI and PII (Table 1). After the second round, this number increased to 8.5 × 105 and 4.3 × 106 cfu/mL for PI and PII, respectively, which implies an increase in the output number of phage after just one round of affinity selection. After five different rounds of panning, the enrichment factor gradually increased from 14 to 3100 for PI and from 58 to 2700 for PII. This was taken as a primary evidence of the successful enrichment of 4-1BB-specific phages.
Enrichment of the anti-4-1BB antibody phages after five rounds of biopanning.

Phage inputs and outputs were determined using the infection of TG1 Escherichia coli and plating on ampicillin plates.
Enrichment factor was determined by dividing the output/input ratio in each round by the ratio in the first round.
To confirm the suitability of the selection procedure, the specificity of the polyclonal phages after each round of panning was verified by ELISA using peptide I and II in MaxiSorp 96-well plates. These data demonstrate that the affinities of phages were increased round by round and the panning procedure has been effective in the enrichment of the 4-1BB-specific scFvs. Elusion phage clones (outputs) from the third, fourth, and fifth rounds were used to assess individual clones. Also, these results confirmed that eluted particles maintained their infectivity properties (Figure 1).
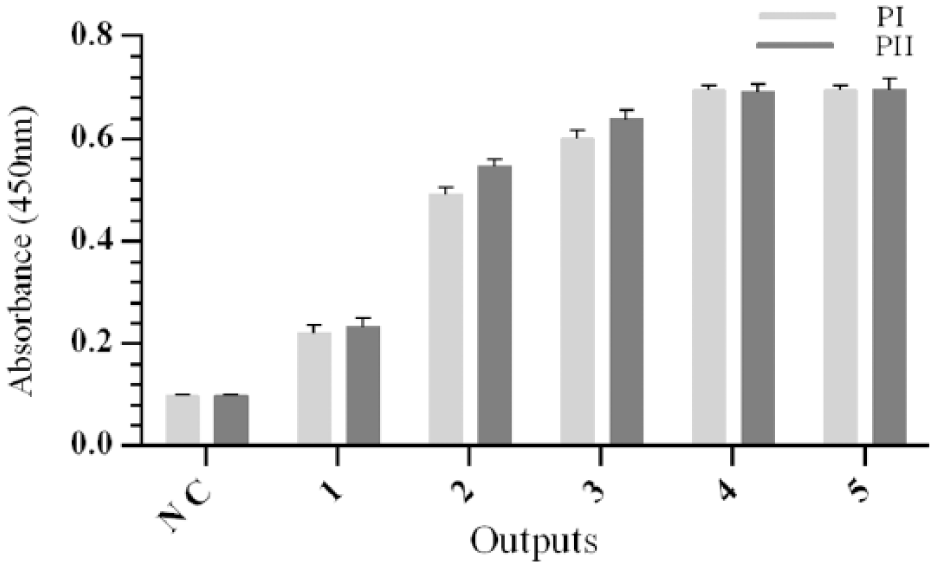
Polyclonal phage ELISA results. Binding activity of amplified phage pools from five rounds of panning on peptides I and II was analyzed by polyclonal phage ELISA. The progressive increase of signal in each round demonstrates that the desired enrichment has been achieved. Data represent the mean of three experiments ± SD.
Screening of individual scFv-phage clones by monoclonal phage ELISA
To isolate and identify the scFvs recognizing peptides I and II, outputs from the third, fourth, and fifth rounds were used. In all, 200 individual phage colonies were picked and their binding activity to peptides I and II was assessed by monoclonal phage ELISA. Out of 200 clones, 57 clones produced phage particles that bound to peptides I and II (27 clones for PI and 30 clones for PII; Figure 2(a) and (b)). Anti-BSA phage (provided with the library) was used as a positive control.

Binding analysis of individual clones in a monoclonal phage ELISA with (a) PI and (b) PII. Phage-infected Escherichia coli TG1 culture supernatants containing approximately 1012/mL phage particles were analyzed in this assay. Clones were considered as positive when the OD in peptides I and II wells was at least three times more than the signal seen in BSA-coated wells. Data represent the mean of three experiments ± SD.
Production and analysis of soluble scFvs
A total of 57 individual scFv clones showing positive signal in monoclonal phage ELISA were used to transfect E. coli Rosetta-gami 2. Unlike TG1, Rosetta-gami 2 is not able to suppress the amber stop codon upstream of the gene III sequence in PIT2 vector, thus resulting in expression of soluble scFv. After IPTG induction and periplasmic extraction, each scFv clone was analyzed by ELISA. The periplasmic extract ELISA data for 57 clones are shown in Figure 3(a) and (b). A total of 40 positive clones were chosen for additional characterization.
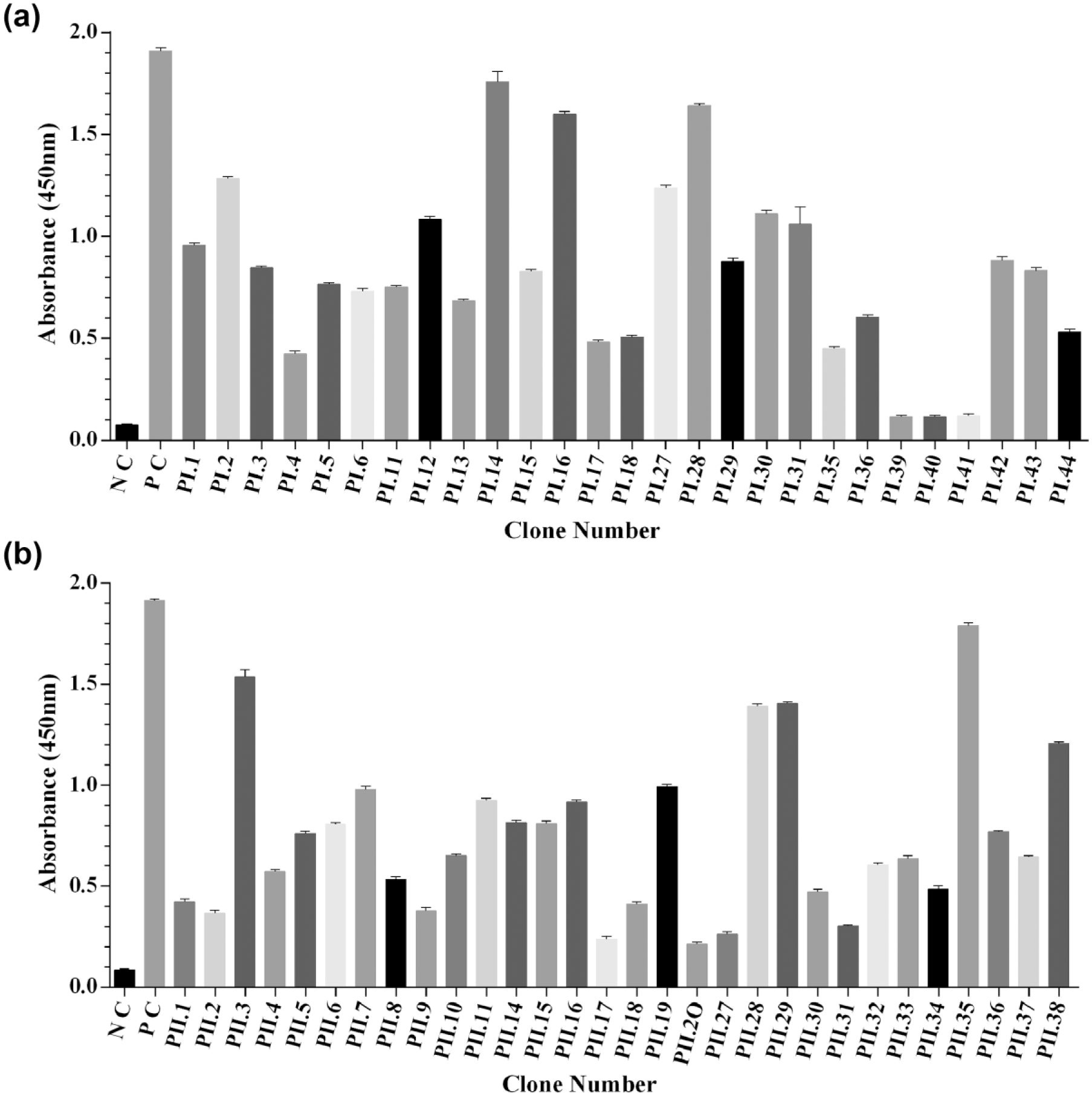
Results of periplasmic extract ELISA. Individual scFv positive clones selected from monoclonal phage ELISA against peptides (a) I and (b) II were used in an ELISA assay. Clones that exhibited at least three times stronger ELISA signals on peptides I and II in comparison to signals on BSA-coated plates were scored as positive. Data represent the mean of three experiments ± SD.
PCR and sequence analysis of the positive clones
A total of 40 positive clones selected from the previous step were amplified by PCR with LMB3 and pHEN primers. Then, the PCR products were analyzed in agarose gel to estimate the proportion of VH–VL insert–bearing phage clones (VH–VL insert, approximately 935 bp). A total of 16 out of the 40 clones analyzed showed a band with the desired size (Figure 4(a)). The positive clones verified by PCR were chosen for additional characterization. DNA fingerprinting with BstNI restriction enzyme showed that some scFvs were different (Figure 4(b)). For further analysis, seven clones with different patterns were sequenced. The framework (FR) and complementary determining regions (CDRs) of the VH and VL chains were determined using the IMGT database. From seven clones nucleotide sequenced, a total of four different sequences were obtained. The amino acid sequences of these selected scFvs are shown in Table 2.

PCR and fingerprinting results. (a) PCR products of selected scFvs; Amplification of scFv fragments by PCR revealed a band of about 935 bp (M: 100 bp DNA marker, other lanes: amplified scFv gene fragments). (b). DNA fingerprinting of scFv clones; scFv gene fragments were amplified by PCR then digested with BstNI enzyme and separated on an agarose gel (M: 100 bp DNA marker, other lanes: single clones digested by BstNI).
The deduced amino acid sequences of anti-4-1BB scFvs selected from phage libraries.

The framework (FW) and complementary determining regions (CDR) were determined using the IMGT database.
Specificity
To assess the specificity of scFvs, the possible cross-reactivity of 4-1BB scFvs with other proteins was investigated by ELISA. Results from this assay indicated that all four scFvs were highly specific for the target antigen and showed no cross-reactivity with other antigens (Table 3).
Specificity of the selected scFvs.

c-Met: hepatocyte growth factor receptor; IGF-1R: insulin-like growth factor 1 receptor; ROR1: receptor tyrosine kinase-like orphan receptor 1; PD1: programmed cell death protein 1; BSA: bovine serum albumin.
Binding was determined by ELISA to a variety of immobilized peptides and proteins. Peptides: 4-1BB receptor (CD137), IGF-1R, c-Met, ROR1; proteins: PD1, skimmed milk, and BSA. The data are presented as mean ± SD from triplicate experiments.
Western blot
Western blot analysis was carried out to verify the correct expression and molecular weight of scFv protein samples using an HRP-conjugated anti-C-myc antibody. The selected scFvs were detected at the expected molecular weight of ~29 kDa which confirmed the successful expression of the soluble scFvs (Figure 5).

Western blotting of anti-4-1BB scFvs. Soluble scFvs were reacted with HRP-conjugated anti-C-myc antibody and detected by 3, 3′-diaminobenzidine (DAB) as substrate. Lane 1: Periplasmic extract of uninduced Escherichia coli Rosetta-gami 2 as negative control, Lane 2: PI.12, Lane 3: PI.42, Lane 4: PII.16, and Lane 5: PII.29.
Flow cytometry analysis for cell binding specificity
In order to evaluate the cell surface binding of the selected scFvs to 4-1BB antigen expressed on the CCRF-CEM cells, a flow cytometry assay was designed. The binding was evaluated by FITC-conjugated anti-M-13 antibody and unstimulated CCRF-CEM cells were included as 4-1BB negative control. No shift in fluorescent intensity was detected in unstimulated CCRF-CEM cells stained with commercial anti-4-1BB IgG (Figure 6). However, based on the shift in fluorescence intensity of isotype control, anti-4-1BB IgG bound to 53% of stimulated CCRF-CEM cells. As shown by the shift in fluorescence intensity value of the cells stained by c-Met as isotype control, four scFvs bound to CCRF-CEM cells: PI.12, PI.42, PII.16, and PII.29 scFvs fragments revealed specific binding of about 19%, 21%, 15%, and 12%, respectively, to CCRF-CEM cells. The fluorescence intensities obtained from unstimulated CCRF-CEM cells stained with the selected scFvs were nearly identical to the value measured from the c-Met treated cells, as isotype control.

FACS analysis of the binding activity of scFvs. The binding activity of the selected scFvs was assessed by flow cytometry 48 h after stimulation with IL-2 and anti-CD3 antibody. CCRF-CEM cells were incubated with scFv and then stained with FITC-conjugated anti-M13 for flow cytometry analysis. Commercial anti-4-1BB IgG antibody and anti-c-Met scFv were used as positive and isotype controls, respectively. The shift in fluorescence intensity was compared to that of isotype control antibody. The four scFvs bound to the CCRF-CEM cells as shown by an increase in the fluorescence value compared to the value measured for the cells treated with anti-c-Met scFv. No shift in fluorescent intensity was observed in anti-4-1BB antibody and scFv treated unstimulated CCRF-CEM cells (Thin line: isotype control, bold line: scFv or commercial anti-4-1BB antibody).
IL-2 mRNA expression in CCRF-CEM cells treated with anti-4-1BB scFvs
The mRNA expression level of IL-2 was evaluated in treated CCRF-CEM cells using real-time PCR. The results indicated that treatment with PI.12, PI.42, and PII.29 scFvs increased the IL-2 mRNA expression (p < 0.05) from 1.00 (±0.13) to 8.79 (±0.14), 9.72 (±0.18), and 12.02(±0.05), in the treated cells, respectively (Figure 7). However, no significant difference of IL-2 mRNA expression was observed in the cells treated with PII.16 and control scFv (ES1).

Relative mRNA expression of IL-2. The mRNA expression was normalized against HPRT. A significant increase of IL-2 was observed in cells treated with PI.12, PI.42, and PII.29 scFvs (p < 0.05). PII.16 and ES1 scFvs could not induce IL-2 production in CCRF-CEM cells.
Induction of IL-2 secretion by anti-4-1BB scFvs
The level of IL-2 secreted by CCRF-CEM cells treated with PI.12, PI.42, PII.16, and PII.29 was measured using ELISA. The results revealed that PI.12, PI.42, and PII.29 caused a significant increase (p < 0.05) in IL-2 production compared with untreated control group (Figure 8). Increased production of IL-2 was observed in cells treated with PII.16, but this evidence was not significant as compared with control. Moreover, ES1 could not induce IL-2 production in CCRF-CEM cells.

IL-2 production by treated CCRF-CEM cells. Cytokine production by CCRF-CEM cells treated with anti-4-1BB scFvs was determined using ELISA. The results are mean ± SD of cytokine secretion level (in pg/mL) for three samples per treatment. PI.12, PI.42, and PII.29 scFvs significantly increased IL-2 production (p ≤ 0.05) but PII.16 could not induce T cells to produce IL-2. ES1 scFv had no effect on IL-2 secretion.
Expression of CD69 on the surface of CCRF-CEM cells stimulated with anti-4-1BB scFvs
The level of CD69 expression on the cells treated with the selected scFvs was analyzed by flow cytometry. In comparing with the control group, PI.12, PI.42, PII.16, and PII.29 scFvs could induce CD69 expression on CCRF-CEM cells (5.8%, 14.7%, 16.9%, 18.1%, and 0.2%, respectively; Figure 9). However, ES1 did not induce CD69 expression.
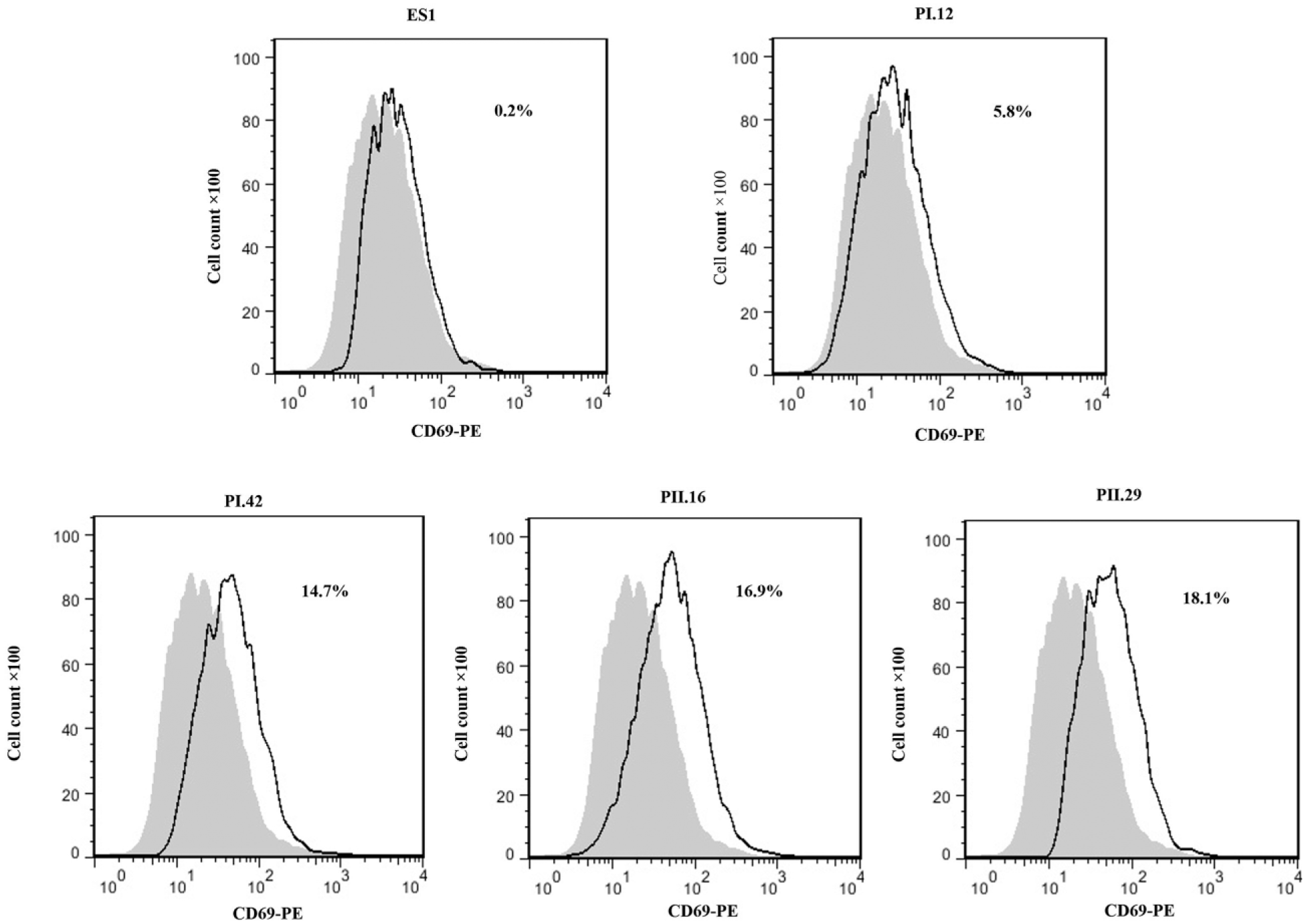
Effect of anti-4-1BB scFvs on the CD69 expression. Expression level of the cell activation marker CD69 was measured on the cells by anti-CD69 PE antibody. The percentage of CD69 expressing cells is shown in histograms. Gray filled histograms indicate untreated control CCRF-CEM cells and dotted line histograms indicate CCRF-CEM cells treated with PI.12, PI.42, PII.16, PII.29, and ES1 scFvs.
Discussion
In humans, costimulation through 4-1BB signaling has been shown to stimulate both CD4 and CD8 T cells, and other immune cells. Moreover, 4-1BB signaling has been described in a wide variety of human disorders including solid tumors, autoimmune diseases, and viral infections. An anti-human 4-1BB mAb has been reported to stimulate T-cell activities.2,8 Furthermore, experimental results have suggested that targeting 4-1BB signaling pathway has a significant role in the therapeutic strategies which has led to investigations on anti-human 4-1BB mAbs as therapeutic agents against human cancers, viral infections, and autoimmune diseases. Therefore, it seems that there is an essential need to develop some antibodies against 4-1BB molecule which may be useful in treatment of the above-mentioned maladies.
Human mAbs against tumor antigens have been considered as suitable targeted therapies for many types of cancer treatments. So far, most of mAbs have been of mouse origin that have limited usage in human clinical trials because of their immunogenicity. Phage display technology developed in recent years provides an alternative to traditional antibody production methods. This technique involves the selection of a number of high-affinity antigen-reactive antibody fragments by employing libraries of recombinant bacteriophages that expose functional antigen binding sites on their surface. Phage display technology is widely used as a method to isolate human scFvs which recognize diverse proteins. Tomlinson I+J are phagemid synthetic libraries of human scFv that are based on a single human framework for VH and VL. The advantage of such libraries is avoiding animal immunization and the possibility to obtain specific scFvs against toxins, allergenic compounds, or self-antigens. Tomlinson I+J libraries have been used in numerous laboratories, because they enable us to obtain human antibodies which are useful for possible therapeutic applications. 24
In recent years, the ability to produce human-derived single chain antibodies has been improved to a point that these small molecules are being used as substitutions for whole mAbs. One of the advantages of scFvs for immunotherapy is their smaller size compared to the mAbs. Moreover, human scFv fragments can penetrate faster and deeper into tissues and they have also been shown to easily clear from the blood, compared to the mAbs or Fabs. Due to the lack of constant regions, scFv retention by Fc receptors found on tissues and organs decreases significantly which in turn can reduce their side effects and make them less immunogenic.
In this study, Tomlinson I+J libraries were panned against synthetic specific epitopes designed from extracellular domain of 4-1BB. We used two specific immunodominant oligopeptides, namely peptide I, consisted of 14 amino acids [TKKGCKDCCFGTFN], and peptide II, composed of 14 amino acids [GTKERDVVCGPSPA]. To enhance the efficiency of biopanning and to reduce the number of the false positive phage clones, we added a pre-absorption step at each round. The phage pools were added to the BSA-coated tubes, and then, the supernatant was transferred to the tubes coated with 4-1BB peptides.
After five rounds of biopanning, the phage titer of the fifth round was much higher than that of the first round (Figure 1). To confirm the suitability of the selection procedure, the specificity of the polyclonal phages after each round of panning was verified by ELISA. According to the previous phage ELISA experiments, a positive reactivity of a selected scFv with a specific peptide occurs when the average of the OD values is at least 2-fold greater than that of the wells without the peptide. A total of 57 clones reacted specifically with 4-1BB and gave strong ELISA signals. However, no signal was observed with the control protein (Figure 2). To examine the production of functional scFvs, periplasmic extract ELISA was performed. Clones that exhibited at least three times stronger ELISA signals on peptides I and II in comparison to signals on BSA-coated plates were scored as positive (Figure 3). PCR analysis of these clones confirmed the presence of the desired band corresponding to the scFv gene. DNA fingerprinting with BstNI restriction enzyme showed that some scFvs were different and revealed common patterns (Figure 4(a) and (b)). By sequencing seven individual clones, we identified four unique anti-4-1BB phage clones (i.e. PI.12, PI.42, PII.16, and PII.29; Table 3). Anti-4-1BB scFvs could successfully bind to 4-1BB antigen expressed on the surface of CCRF-CEM cells (Figure 6) confirming the functionality of the selected scFvs. Then, the stimulatory effect of anti-4-1BB scFvs was investigated in CCRF-CEM cells. It was shown that cell treatment with anti-4-1BB scFvs induced IL-2 mRNA expression and IL-2 protein production (Figures 7 and 8), and also enhanced expression of CD69 on the surface of CCRF-CEM cells (Figure 9).
The aim this study was to select some scFvs against human 4-1BB antigen from Tomlinson I+J libraries. The main conclusion to be drawn from this study is that PI.12, PI.42, PII.16, and PII.29 scFvs have in vitro targeting property that is well suited for 4-1BB detection. We will use these scFvs that showed an acceptable binding to the target cells for the functional studies. Also, results suggest that scFvs were able to affect CCRF-CEM immune responses, which provides evidence for immunomodulation advantages of scFvs. Further investigation needs to be done to establish whether selected scFvs have in vitro and in vivo efficacies to act as anti-tumor or anti-viral agents.
Footnotes
Acknowledgements
The authors thank Dr Soheila Ajdary for helping them with the western blot and flow cytometry experiments and also Dr Fariborz Bahrami for his help with the PCR experiments and bioinformatics analysis.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by the Pasteur Institute of Iran (Grant No. 818).