
Abstract
Proteasome is a multi-protein organelle that participates in cellular proteostasis by destroying damaged or short-lived proteins in an organized manner guided by the ubiquitination signal. By being in a central place in the cellular protein complement homeostasis, proteasome is involved in virtually all cell processes including decisions on cell survival or death, cell cycle, and differentiation. These processes are important also in cancer, and thus, the proteasome is an important regulator of carcinogenesis. Cancers include a variety of cells which, according to the cancer stem cell theory, descend from a small percentage of cancer stem cells, alternatively termed tumor-initiating cells. These cells constitute the subsets that have the ability to propagate the whole variety of cancer and repopulate tumors after cytostatic therapies. Proteasome plays a role in cellular processes in cancer stem cells, but it has been found to have a decreased function in them compared to the rest of cancer cells. This article will discuss the transcriptional regulation of proteasome sub-unit proteins in cancer and in particular cancer stem cells and the relationship of the proteasome with the pluripotency that is the defining characteristic of stem cells. Therapeutic opportunities that present from the understanding of the proteasome role will also be discussed.
Introduction: cancer stem cell theory
Normal adult tissues rely on a small percentage of stem cells that retain the potential to self-renew and differentiate toward cell types of their resident organ and serve in cell replacement and organ regeneration during the adult organism lifespan. 1 Such stem cells exist in several organs including the normal human skin, breast, bowel, and other organs.2,3 Cancer stem cells (CSCs or tumor-initiating cells) are the neoplastic counterparts of normal adult tissue resident stem cells and represent usually a small percentage of the total cancer cell population. The CSC hypothesis stipulates that they are the most important contributors in tumor development and propagation. The origin of CSCs is debatable, and they may be derived from malignant transformation of normal tissue stem cells or from transformation of differentiated cells that acquire mutations allowing dedifferentiation and acquisition of tumor-initiating properties. Mutations in genes important for determination of cell fate are prominent in malignancies. 4 Tumor-initiating properties are defined experimentally and include the ability of cells to be serially passaged in immunosuppressed mice and produce tumors with the heterogeneity of cells observed in the initial tumor, even when transplanted in mice at much lower numbers than the bulk of tumor cells.5,6 Both derivations of tumor stem cells from either normal stem cells or differentiated cells could be operative in different patients, and the two scenarios are not mutually exclusive. Variable routes of establishment of CSCs may in fact explain the different sub-types of cancers seen in the same organ, as neoplastic stem cells may retain epigenetic memory of their initial cell of provenance and re-differentiate back when producing the bulk cancer cell population. In breast cancer, it has been experimentally confirmed that about 200 stem cells with the CD44high/CD24low phenotype can reconstitute tumors when implanted in mice while more than 50 × 103 of unsorted, bulk cancer cells are needed. 5 Similarly, CSCs with cancer-initiating properties and partially overlapping surface markers have been identified in colon cancer, leukemia, and other cancers.7–9
CSCs contain mutations that lead to the acquisition of additional properties on top of the tumor-initiating cells’ properties described above which are essentially properties of normal tissue stem cells. These mutations endow them with the required abilities for cancer development and maintenance, including sustained proliferation and evasion from growth suppressors, resistance to apoptosis, induction of angiogenesis, invasion and metastasis potential, genome instability, avoidance of immune recognition, and destruction and metabolism adjustments. 10
The proteasome is a multi-protein cellular complex situated in both the cytoplasm and nucleus and involved in protein degradation. Proteasome sub-units and their expression in cancer will be examined in detail in this article.
Proteasome structure and function
The ubiquitin proteasome system (UPS) is a multi-protein machinery that serves in the degradation of defective or unwanted proteins. This positioning in the center of proteostasis makes the UPS an important regulator of cellular metabolism in general. A cascade of three types of enzymes known as E1 (or ubiquitin-activating enzyme), E2 (or ubiquitin-conjugating enzymes), and E3 (or ubiquitin ligases) attach the small protein ubiquitin to a substrate protein that is then recognized and degraded by the proteasome particle 11 (Figure 1). The UPS handles hundreds of different proteins and thus participates in the regulation of virtually every cellular process. It has been shown to participate in pluripotency induction as well as the reverse process of differentiation. 12 UPS is also involved in carcinogenesis, and inhibitors of the proteasome are in use for a number of years in the treatment of hematologic cancers. 13 Thus, regulation of proteasome in CSCs may have important implications for carcinogenesis and cancer therapeutics.

The ATP-dependent ubiquitination cascade leading to delivery of target proteins to the proteasome for degradation. The 26S proteasome with its different components is depicted in the lower part of the figure. E1: ubiquitin-activating enzyme; E2: ubiquitin-conjugating enzyme; E3: ubiquitin ligase.
The proteasome (26S proteasome) is a hollow cylinder–shaped multi-protein structure of 2.5 MDa which comprises a core particle (CP or 20S proteasome) covered in one or both sides by a regulatory particle (RP or 19S proteasome; Figure 1). RP comprises a lid and a base sub-complex and functions in ubiquitinated proteins recognition, de-ubiquitination which allows ubiquitin molecules to be recycled, unfolding of the target proteins, and delivery of the proteins to the CP. 11 The different sub-units of RP possess specific activities to accomplish all these functions. Three sub-units of the base sub-complex, PSMD4 (also called Rpn10 or S5a), Rpn13 (alternatively called ADRM1), and PSMD10 (or Gankyrin), possess ubiquitin recognition domains that allow them to recognize poly-ubiquitin chains. Sub-unit PSMD14 (alternatively named S13 or Rpn11) of the lid sub-complex is a de-ubiquitinatinase and recycles ubiquitin from proteins that had been recognized. The 19S base sub-complex is made up of six ATPases and three other peptides without ATPase activity. ATPases belong to the AAA (ATPases Associated with various cellular Activities) family and are able to hydrolyze all four nucleotide triphosphates and to alter the conformation of proteasome substrate protein, preventing their aggregation before they enter the CP to be degraded. 11 Sub-unit PSMD11 (S8 or Rpn6) helps consolidate the stability of the CP–RP interaction by making conducts at the level of the ATPase unit PSMC5 (Rpt6) and the α ring component PSMA2 (α2). 14
CP is made of four rings of seven member proteins each that are stacked one on the other. The two identical peripheral rings are called α rings (with sub-units PSMA1–7 also called α1–α7), and the two also identical central rings are called β rings (with sub-units PSMB1–7 also called β1–β7, although the two nomenclatures are not numbered in parallel, for example, enzymatic sub-unit β1 is PSMB6, β2 is PSMB7, and only for the third enzymatic sub-unit, the two nomenclatures attribute the same number, β5 being PSMB5). 15 The proteasome possess three enzymatic activities, a post-glutamyl (caspase-like or post-acidic residues cleavage) activity, a trypsin-like (post-basic residues cleavage) activity, and a chymotrypsin-like (post-hydrophobic residues cleavage) activity that reside in sub-units PSMB6, PSMB7, and PSMB5 (β1, β2, and β5), respectively, and degrade target proteins producing fragments of four to 14 amino acids. 16 α rings that flank the two β rings function to prevent proteins from being degraded randomly. This is accomplished by obstructing access to the β rings that possess the enzymatic activity. Interaction of the α units with the 19S RP leads to open configuration of the α ring which allows targeted proteins to pass. Thus, the proteasome is active when fully assembled, while the 20S particle when not associated with one or two RPs has minimal activity, mainly toward small proteins that can enter it without being ubiquitinated. 17
Alternative enzymatic sub-units may replace the enzymatically active β ring sub-units giving rise to immunoproteasome. These sub-units designated PSMB8 (also called β5i or LMP7), PSMB9 (also called β1i or LMP2), and PSMB10 (also called β2i or MECL-1, multicatalytic endopeptidase complex-like 1) have modified enzymatic activities compared with the regular proteasome sub-units and favor generation of fragments that are presented by major histocompatibility complex (MHC) class I receptors for antibody generation. In addition, immunoproteasome is expressed in tissues beyond immune cells and may have functions beyond immune presentation. 18 It is possible also, in some occasions depending on sub-unit availability, for α rings to assemble in a non-stoichiometric fashion. Proteasomes with two PSMA7 sub-units and no PSMA4, for example, have been observed in mammalian cells. 19
Assembly of the proteasome complex is an ordered process and is facilitated by proteins that are not part of the proteasome structure but participate in the seamless progression of the process. 20 Five factors have been confirmed to participate in the assembly of 20S CP, which starts with the assembly of α ring. These facilitators are the four factors PSMG1–4 (also known as PAC1–4) and protein POMP (proteasome maturation protein also called proteassemblin or UMP1). β ring is then assembled on the α ring, creating a half proteasome which then interacts with another nascent half proteasome to create the full 20S. 20 POMP is recycled during the process, while PSMG1–4 factors are degraded. The details of the 19S RP assembly are less well outlined, but it appears that the 20S proteasome plays a role in this assembly. 20 Other factors such as valosin-containing protein (VCP) contribute to the assembly of 19S with 20S to form the 26S proteasome. 21
Instead of the 19S, alternative RPs may associate with the 20S proteasome. These include the 11S (PA28) which is a heteroeptamer comprising three PSME1 (also called PA28α) and four PSME2 (also called PA28β) units and is mostly assembled with the immunoproteasome. 22 Other alternative RPs include PA28γ (PSME3) and PA200 (PSME4). All these alternative proteasome activators lack ATPase activity and do not recognize ubiquitin. 23 As a result, they are able to target small proteins with simple tertiary structures such as the cyclin dependent kinase (CDK) inhibitors p16, p19, and p21. Hybrid proteasomes with one 19S RP and one 11S flanking the CP exist and may function in polypeptide production for immune presentation with RP as the site of protein entry and 11S as the side of exit that facilitates the production of peptides with the appropriate size for presentation. 24
The function of the proteasome is versatile, and besides complete destruction of targeted proteins, it may process target proteins only partially in a regulated manner which may lead to functional activation instead of permanent disposal. A well-described example of partial proteasomal degradation is observed in the case of the activation of the alternative NF-κB pathway after partial cutting of factor p100. 25
Both structural and assembling proteasome proteins are important for the overall function of the organelle, and even dysregulation of one component may have global implications for this function as will be discussed in subsequent sections.
Proteasome activity in cancer and CSCs
Perturbations of proteasome activity in CSCs are of interest because altered activity may be associated with not only the pathobiology of these cells but also therapeutic opportunities. Moreover, comparison of this activity between CSCs and the rest of the tumor cells as well as the normal tissue stem cells and normal embryonic cells may reveal differences in pathways that could be further exploited therapeutically.
A study with glioblastoma and breast carcinoma cells engineered to express a proteasome substrate fused to a green fluorescent protein (in order to be tracked down in vivo) showed that cells growing in sphere cultures, enriching for CSCs and progenitor cells, had lower proteasome activity than cells growing in monolayers. 26 In this system, cells with low proteasome activity express the green fluorescent protein. In a modification of the transfection construct, a thymidine kinase sequence was added in order to be able to pharmacologically target cells having low proteasome activity and stably express the construct. 27 Efficient regression of xenotransplants established from breast cancer cell lines T47D and MDA-MB-231 sorted for low proteasome activity in mice was shown by treatment with ganciclovir, activated by co-expressed thymidine kinase. 27 By using the same proteasome substrate fused to a green fluorescent protein system transfected in osteosarcoma cell lines, another group showed that cells with low proteasome activity (positive for the green fluorescence) were able to divide asymmetrically and produce both cells with high and low proteasome activity while cells with higher proteasome activity (green fluorescence negative) could produce only green fluorescence–negative progeny 28 (Figure 2). In addition, the study confirmed that cells with low proteasome activity had a higher sphere-forming capacity than high proteasome activity counterparts. Nevertheless, neither sub-group of osteosarcoma cells could establish xenografts in nude mice after up to 105 cells per mouse have been inoculated, a fact attributed to inability to inoculate higher numbers due to technical reasons, 28 but that, in fact, may imply that low proteasome activity cells are heterogeneous and only a percentage among them are true stem or tumor-initiating cells.

Asymmetric cell divisions in cancer stem cells (CSCs) with low proteasome activity give rise to a similar CSC with low proteasome activity and a daughter cell destined to differentiate and which has (or will obtain along the way) high proteasome activity. As cancer progresses, CSCs undergo also symmetric divisions that increase the number of stem cells with low proteasome activity.
Human head and neck cancer cell lines treated with radiation became enriched in low proteasome activity cells, supporting the view that CSCs and progenitor cells are responsible for tumor radioresistance. 29 The low proteasome activity fraction had higher frequency of cells with tumor-initiating capability in mice. A tumor microarray investigation disclosed that patients with head and neck cancer whose tumors had a lower expression of the 19S proteasome sub-unit PSMD1 had worse overall survival than patients with higher expression of PSMD1. 29 Similarly to head and neck cancer cells, non-small-cell cancer cell lines growing as spheres had lower proteasome activity than monolayers and were more tumorigenic in mice. 30 Additionally, prostate cancer cell line fractions with low proteasome activity were more radioresistant than the high proteasome activity fraction. 31 Another group used a lentiviral transfection system to label a fluorescent proteasome activity marker in human breast cancer cells and confirmed that cells with low proteasome activity injected orthotopically in mammary pads of immunocompromised mice produced larger tumors and more metastatic spreads than high proteasome activity counterparts. 32 A similar result was obtained when mouse breast cancer cells were injected directly into the circulation of immunocompetent mice, confirming that the results with human cells are valid even when immune surveillance is intact and additionally confirming that the metastatic activity of human low proteasome cells is independent of primary tumor site. Primary human glioblastoma cells with low proteasome activity produced tumors in mice more often than high-activity counterparts when only 5 × 103 cells were injected. 32 Low proteasome activity cells had a higher expression of the stem cell marker and pluripotency-associated transcription factor Sox2. In addition, human unsorted glioblastoma cell line xenografts were resistant to the proteasome inhibitor NPI-0052 (salinosporin A), consistent with failure of the drug to eradicate stem cell populations. 33 Another study showed that colon cancer cell fractions with stem cell properties, such as increased sphere formation capacity, chemo- and radio-resistance, and ability to engraft and produce tumors when xenografted in nude mice in low numbers, displayed low proteasome activity. 34
Based on the results of decreased proteasome activity in CSCs, another study took a reverse approach and sought to establish whether proteasome inhibition would promote the stem cell phenotype. 35 Inhibition of β2 or β5 but not β1 enzymatic activity in breast cancer cells increased expression of CD44, a stem cell marker in these cells, and also promoted epithelial–mesenchymal transition (EMT) phenotype. Overall expression of proteasome units was not affected by the pharmacologic enzymatic inhibition applied experimentally. In addition, patients with breast cancer with low PSMB2 and PSMB5 units expression in their tumor tissue had a worse survival than high-expression counterparts. 35 Of note, though, the PSMB2 unit checked in this study through the genomic Oncomine platform would correspond to a non-enzymatic unit of the β ring (β4) and not the intended β2 unit.
Decreased proteasome activity in CSCs may have important implications for the protection of these cells from host immune attack by decreased presentation of tumor neo-antigens. Indeed, agreeing with this hypothesis, a polymorphism substituting glutamine for lysine in position 49 of the pro-sequence of PSMB8 (LMP7) unit, which leads to failure of the protein to be induced by interferon-γ (IFN-γ), was more prevalent in colon cancer patients than in healthy controls, suggesting that the failure of induction affects the ability of the immune system to mount an anti-cancer response. 36
Overall, these investigations associate low proteasome activity with populations of CSCs and with the ability to propagate tumors in vivo and resist treatment. The down-regulation of proteasome activity may be a result of the low metabolic activity of stem cells that is associated with low cell cycling. Moreover, it may be a prerequisite for the stability of the proteins of the pluripotency network, several of which are proteasome target proteins. 37 In contrast, studies of proteasome expression and activity in the bulk carcinoma cells have shown increased activity and expression in several carcinomas. A genomic study using publicly available genomic databases showed several proteasome sub-units, including PSMB3, PSMA4, PSMD4, PSMA5, PSMD2, PSMA6, PSMA2, and PSMB7, to be up-regulated compared to normal tissues. 38 Another study that examined UPS activity in breast cancer tissue confirmed increased activity compared with adjacent normal tissues. 39 PSMC2 (Rpt1 in yeast) expression was examined as representative of proteasome sub-units and was found to be increased together with other UPS components including E3 ligases. Similarly, increased expression and activity was evident in serous ovarian carcinoma tissues and cell lines compared to benign tumors or immortalized ovarian surface epithelium. 40 Despite this increase, a concomitant increase in ubiquitinated substrates and sensitivity to pharmacologic proteasome inhibition was observed, implying that the increased proteasomal activity is inadequate to satisfy the demands of proteostasis in neoplastic cells. In a genomic analysis of differentially expressed genes between bladder cancer and normal bladder tissues, β ring component PSMB1 was one of the top up-regulated genes. 41 19S lid component PSMD11 was among the top down-regulated proteins after PI3K inhibition in lung and pancreatic cancer cells in another proteomic study. 42
Consistent with the association of low cycling and low proteasome activity, normal human embryonic stem cells (hESCs), which, in contrast to CSC counterparts, have a high cycling activity during developmental organogenesis, possess a high proteasome activity. 43 This is an important distinction to emphasize with pathogenic and possible therapeutic implications. Proteasome activity in these cells is an inbuilt property with pluripotency, given that proteasome inhibition by exposure to the inhibitor MG-132 promotes differentiation. Several proteasome sub-units show a decrease in their level of expression after differentiation, with the alternative immunoproteasome sub-units PSMB8 and PSMB9 exhibiting the most striking reduction. 44 Not only proteasome activity is required for the maintenance of pluripotency in ESCs and pluripotency induction in induced pluripotent stem cells (iPSCs) but also components of the ubiquitin–proteasome cascade are prerequisites for induction of differentiation. 45 High proteasome activity is dependent on the transcription factor FOXO4 up-regulation of the 19S sub-unit PSMD11. 43 Thus, it seems that high proteasome activity in normal hESCs maintains their pluripotency and also contributes to their ability to differentiate toward different lineages by keeping differentiation genes inactive but poised for activation. 46 Interestingly, in murine ESCs, induction of the alternative activator PA28 is observed just after induction of differentiation. 47 This induction would promote ubiquitination-independent elimination of damaged proteins, but has not been seen in human cells, introducing the possibility that there are species differences in proteasome regulation during development and possibly more generally. 22 Sub-unit NF-YA of transcription factor NF-Y (nuclear factor Y), which, as it will be discussed in the following section, up-regulates proteasome sub-units, is involved in positive regulation of cycle regulator CDCA8 in embryonic cells, and thus, a program of proteasome up-regulation, satisfying increased metabolic needs, may be coordinately regulated with the cell cycle. 48 In addition, a short isoform of NF-YA regulates the pluripotency program itself in embryonic cells. 49 This prerequisite of high proteasome activity for gene priming is not shared by normal adult tissue stem cells and CSCs which retain a more restricted ability to differentiate to different cells of tissue of origin or to different bulk cancer cells (Figure 3). Interestingly, senescent cells during the aging process display a decreased proteasome activity which may be involved in the association of cancer and aging. 50 Figure 3 provides a schematic comparison of proteasome activity differences in cancer, normal adult tissues, and embryonic development, as discussed in this section.

Schematic representation of proteasome activity in cancer (upper graph), adult tissues (middle graph), and development (lower graph) as cells progress from cancer stem cells (CSCs) to differentiated cancer cells, from tissue stem cells to senescent cells, and from embryonic stem cells (ESCs) to differentiated embryonic layers, respectively.
Regulation of proteasome sub-units expression
Decreased proteasome activity in CSCs from various cancers may be due, at least in part, to decreased transcription and expression of different proteasome sub-units leading to defects in proteasome assembly, associated with missing constituent parts. The expression of proteasome sub-units is regulated by several transcription factors including NF-Y, NRF1 and NRF2, HSF2, and FOXO factors, acting in different instances after receiving up-stream signals. Some of them may regulate mostly baseline expression while others may have a more prominent role in response to specific stress conditions, although the lines of distinction are not always clear.
NF-Y is a tripartite DNA-binding transcription factor (TF) consisting of sub-units NF-YA, NF-YB, and NF-YC that recognizes the core DNA sequence CCAAT and ATTGG in the complementary chain (Figure 4a). Although NF-Y is not sufficient to induce promoters by itself, it is necessary for such activation, in co-operation with other transcription factors. 51 Examples of co-operating transcription factors include FOXO family factors (see later) and the estrogen receptor alpha (ERα) in breast cancer cells. 52 NF-Y induces the expression of 20S sub-units PSMA2, PSMA3, PSMA5, and PSMB3 but not PSMD3 in vitro. 53 PSMD10 is also a target of NF-Y, and its expression is enhanced by interleukin 1β (IL-1β) signaling in hepatocellular carcinoma. 54 Several other proteasome sub-units including PSMB1, PSMB2, PSMC2, PSMC3, PSMC5, PSMD4, and PSMD14 possess a CCAAT box in their promoters and may be similarly regulated by NF-Y. 53 In contrast, transcription factor C/EBP (CCAAT/enhancer binding protein) that binds the same sequence could not bind the PSMA5 promoter, despite the presence of target sequences, arguing for the importance of additional prerequisites, such as co-operating transcription factors or post-translational modifications, for this binding, enabling NF-Y binding but absent in the case of C/EBP. 53 Down-regulation of NF-Y in stem cells at the post-transcriptional level by the mRNA-binding factor Musashi leads to a decrease in the expression of proteasome sub-units that are targets of NF-Y and decreased proteasome activity in glioblastoma and breast cancer–initiating cells. 55 Decreased proteasome activity results in stabilization of the intra-cellular domain of Notch (Notch-ICD), a transcription regulator with an important role in stem cell signaling (Figure 5). Musashi increases Notch activity by an additional mechanism, involving binding and inhibiting translation of the mRNA encoding for the protein Numb, a negative regulator of Notch. 55 Numb plays additionally roles in asymmetric division and p53 regulation. 56 Thus, its inhibition in stem cells by Musashi would lead to promotion of symmetric divisions, thus increasing the number of cells with CSC properties. 57 However, Musashi down-regulates the mRNA of Notch ligand Jagged1, an action that contributes to the maintenance of epithelial cell identity. 58 Musashi is present in stem cells but not in differentiated cells of various tissues, and thus, NF-Y may become up-regulated upon differentiation. 59 Musashi expression is up-regulated by Notch signaling, at least in colorectal cancer cells, 60 and thus, an intricate feedback loop is in place that results in NF-Y regulation. In addition to availability of NF-Y itself, availability of co-operating transcription factors may be important for proteasome units expression in stem cells. In breast cancer, for example, stem cells of estrogen receptor (ER)–positive cancers do not express ER, and thus, NF-Y would be expected to be functionally impaired even if expressed, in contrast to the rest of the tumor cells, where the expression of ER would enable NF-Y-dependent transcription of proteasome sub-units. 61 Moreover, a feedback loop is also present in this case, as proteasome inhibition suppresses ER expression, a fact that may contribute to ER negativity of breast CSCs 62 (Figure 5).

Logos of consensus DNA-binding sites of the transcription factors with a role in proteasome sub-units genes regulation as listed in the JASPAR database (http://jaspar.genereg.net): (a) NF-YA (H. Sapiens) MA0060.2, (b) MAF:NFE2 (H. sapiens) MA0501.1, (c) HSF2 (H. sapiens) MA0770.1, (d) FOXO4 (H. sapiens) MA0848.1, and (e) STAT3 (H. sapiens) MA0144.2.

Schematic representation of transcription factor NF-Y regulation by Musashi in cancer stem cells (CSCs) on the left and in progeny cancer cells on the right. In CSCs, the presence of Musashi inhibits translation of mRNA of NF-Y along with other mRNAs of Numb and Jagged. Down-regulation of NF-Y along with absence of ER in breast CSCs (or other co-operating factors in other cancers) leads to proteasome sub-units down-regulation. In parallel, Numb down-regulation leads to p53 down-regulation and promotion of symmetric divisions producing more CSCs. Notch is stabilized and may receive signals from neighboring cells. In cancer bulk progeny cells (right), the absence of Musashi leads to NF-Y mRNA stabilization and in the presence of collaborating factors (e.g. ER in breast cancer) up-regulation of proteasome sub-units. mRNAs are depicted as ovals and proteins as blocks. Broken outlines denote down-regulated proteins.
Transcription factors, nuclear factor erythroid-derived 2-related factor 1 and 2 (NRF1 and NRF2), bind DNA elements with the sequence TGAG/CNNNGC known as anti-oxidant response elements (AREs; Figure 4b). They bind as heterodimers with Jun factors (c-Jun, Jun-D, or Jun-B) or musculoaponeurotic fibrosarcoma (MAF) factors. 63 NRF1, also known as NFE2L1, is a basic leucine zipper transcription factor of the cap‘n’collar family transcribed from human chromosome 17q21. NRF1 mediates recovery of proteasome activity after proteasome inhibition in mouse embryonic fibroblasts. 64 The effect is dependent on the presence of AREs in the promoters of proteasome components genes. Most proteasome proteins–encoding genes possess AREs, and some of them, including PSMA2/α2, PSMB4/β7, PSMB6/β1, and PSMC4/S6b, are confirmed to be up-regulated by TCF11, a long isoform of NRF1. 65 Proteasome up-regulation was dependent on the availability of TCF11 in the endoplasmic reticulum before activation which was dependent, in its turn, on the function of insertion factor Sec61α. Interestingly, in this study, NRF2 could not up-regulate these genes. 65 NRF1 is up-regulated down-stream of mammalian target of rapamycin complex 1 (mTORC1) activation. 66 This activation results from increased input from several growth factor pathways and provides amino acids derived from proteins degradation in the proteasome for rebuilding of new proteins mediated by the increased translation due to mTORC1 activity. In contrast, mTORC1 inhibition by mTOR inhibitors rapamycin or torin1 did not lead to proteasome down-regulation. 67 Proteasome expression level was unaltered, and proteasome degradation was in fact increased due to increased ubiquitination of target proteins. These results would argue for alternative regulators contributing to proteasome sub-units expression maintenance after mTOR inhibition that would decrease NRF1, at least in some cellular environments.
NRF2, also known as NFE2L2, is a similar basic leucine zipper transcription factor transcribed from human chromosome 2q31. It also binds AREs in promoters and is activated down-stream of growth factors through the PI3K/Akt cascade which inhibits the NRF2-inhibiting kinase GSK3β. 68 NRF2 up-regulates activity of proteasome in hESCs and co-localizes with OCT4 and NANOG, which are core transcription factors of the pluripotency stem cell program. 69 The regulation of proteasome activity is mediated through the chaperone protein POMP (proteasome maturation protein). Proteasome inhibition promoted differentiation toward mesendoderm by interfering with levels of OCT4 and NANOG. 69 In contrast to ESCs, human senescent fibroblasts have decreased NRF2 function associated with low proteasome activity. 70 Activation of NRF2 in cultures of terminally senescent fibroblasts could still prolong their lifespan. In addition, NRF2 is implicated in reactive oxygen species (ROS) detoxification by up-regulating anti-oxidant enzymes. 71 Exogenous anti-oxidants such as the cruciferous vegetable ingredients sulforaphane and 3H-1,2-dithiole-3-thione promote ROS detoxification by inducing NRF2, and this induction leads to proteasome up-regulation through increased transcription of sub-units such as PSMB5. 72 PSMB5 was also up-regulated by oxidative stress in murine embryonic fibroblasts in a NRF2-dependent manner. 73 In these cells, the immunoproteasome was not induced following oxidative stress. 73 Cancer cells exposed to chemotherapy display a higher percentage of resistant CSC-like fractions that express higher NRF2 levels due to UPS inhibition and stabilization of the NRF2 protein. 74 Moreover, these cells display low levels of ROS. Importantly, NRF2 expression was required for the maintenance of glioblastoma stem cells, and knock-down of NRF2 with short hairpin RNA (shRNA) decreased the ability of human glioblastoma cells to form xenografts in nude mice. 75 In addition, glioblastoma stem cells displayed decreased proliferation and decreased expression of stem cell proteins Sox2 and BMI-1 after NRF2 knock-down.
In contrast to the above data, another study failed to observe changes in the expression of proteasome sub-unit PSMA4/α3 or induction of NRF2 in human fibroblasts that were exposed to heat shock and displayed decreased proteasome activity. 76 This result argues for the presence of a different pathway effectuating proteasome down-regulation in these conditions and underscores the fact that this down-regulation may result from multiple different signals, affecting multiple regulators and multiple processes during both translation and post-translation, and is dependent on cellular context (e.g. CSCs vs fibroblasts). Together with the results described in the previous paragraph and the low proteasome activity of CSCs (as described in the previous section), it appears that NRF2 and proteasome activity are dissociated in CSCs. High levels of NRF2 are not able to maintain proteasome activity in these cells but are required for ROS detoxification (Figure 6). However, low proteasome activity contributes to maintaining NRF2 active, given that NRF2 is a substrate for proteasome degradation after association with inhibitor INrf2 (also called Keap1) and ubiquitination by the ubiquitin ligase Cul3/Rbx1. 77 This provides advantage to tumors by increasing their resistance to oxidative stress and various treatments and is underlined by the fact that both NRF2 and Inrf2 harbor mutations in a minority of cancers.78,79 In contrast, in normal cells, NRF2 acts as a tumor suppressor by inducing anti-oxidant machinery and maintaining proteostasis under stress conditions. 80 Both NRFs appear to have a role in proteasome regulation mostly under stress conditions, when growth factor cascades are activated in a persistent, protracted, un-regulated manner, while the basal proteasome units expression is regulated by other factors such as NF-Y, in the presence of co-operating factors, and FOXOs.

Two hats for transcription factors NRF1 and NRF2. In normal cells (upper schematic), these factors act as tumor suppressors by inducing proteasome and anti-oxidant enzymes. In established cancer cells (lower schematic), these same actions promote cells resistance to environmental stresses and therapies. In cancer stem cells, additional regulators may suppress proteasome expression (not shown).
Heat shock factor 2 (HSF2), a transcription factor regulating the cellular heat shock response, also regulates proteasome sub-units expression.81,82 It often binds DNA as a heterodimer or heterotrimer with the related factor HSF1 on heat shock response sequences (Figure 4c). Cellular heat shock response is activated after growth factor stimulation and oncogene activation. 83 In mouse embryonic fibroblasts, knock-down of HSF2 led to a decreased expression of several 20S sub-units including PSMA1, PSMA5, PSMB2, and PSMB5 as well as 19S sub-units PSMC4 and PSMD10. Other sub-units such as PSMB4, PSMC5, and PSMD4 were affected less or not at all. 81 Decreased HSF2 expression is associated with adverse outcomes in prostate cancer and several other carcinomas such as breast, serous ovarian, lung, and renal. 84 In a detailed analysis of cellular processes, HSF2 suppression was predominantly connected with dysregulation of small GTPase activity leading to altered actin cytoskeleton dynamics and promotion of EMT and metastasis. 84 Interestingly, EMT properties are prominent in CSCs,85,86 which display decreased proteasome activity. Thus, HSF2 suppression may be an event that leads both to decreased proteasome activity associated with pluripotency and to promotion of EMT-associated mobility. Other factors, such as Musashi, provide additional links of low proteasome activity with symmetric divisions and pluripotency potential, both characterizing CSCs.
FOXO4 is a transcription factor of the forkhead box family and, as previously discussed, specifically up-regulates proteasome sub-unit PSMD11 in human ESCs, which display a high proteasome activity associated with diminished replicative senescence and unlimited survival in cell culture. 87 FOXO worm homolog Dauer Formation-16 (DAF-16) promotes proteasome activity and prolongs longevity in Caenorhabditis elegans. 87 In contrast, knock-down of FOXO4 in hESCs interferes with the ability of these cells to differentiate toward neural lineages but not mesendodermal differentiation. 88 These results are similar to the effects of proteasome inhibition mediated by the decrease in NRF2 activity, discussed above, and confirm the role of proteasome in pluripotency of embryonic cells. 69 Both FOXO4 and NRF2 are regulated by the PI3K–Akt pathway but in opposite directions, NRF2 being activated by Akt indirectly through inhibition of GSK3β, while FOXO4 is directly phosphorylated and inhibited 88 (Figure 7). A fine balance between pathway activity and down-stream transcription factors is at play, maintaining the appropriate proteasome activity not only for stem cell function but also during differentiation. This balance is reminiscent of other tight regulations in ESCs such as the levels of pluripotency transcription factors that need to be within a given range in order for pluripotency to be established and maintained while both lower and higher levels favor differentiation. 89 In fact, FOXO family transcription factors have been implicated in the long-term maintenance of hematopoietic stem cells in a quiescent state while their knock-down promotes tumors in mice. 90 FOXOs suppression associated with carcinogenesis promotion would concomitantly lead to the observed decreased proteasome activity in CSCs (Figure 8).

Regulation of several transcription factors with a role in proteasome sub-units transcription by growth factor pathways. The PI3K/Akt/mTOR cascade is located in the center of these regulations and is part also of the general regulation of cellular proteostasis as it participates in autophagy regulation and metabolism.
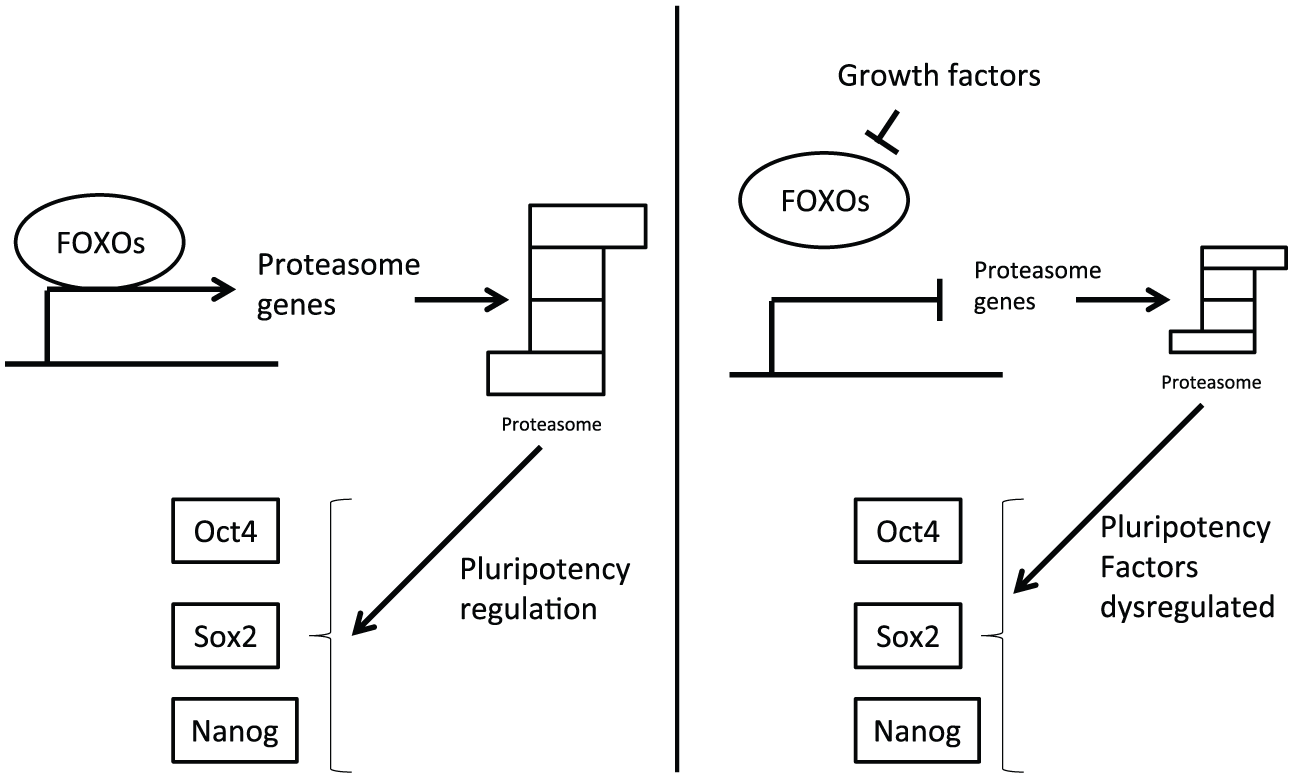
FOXO transcription factors regulation of proteasome (left) is affected by their suppression down-stream of growth factor activation (right). This dysregulation perturbs the pluripotency network core transcription factor balance and associates proteasome function and cancer stem cells. Proteasome down-regulation observed in cancer stem cells may lead to altered levels of the core pluripotency transcription network favoring neoplastic transformation.
Another FOXO family member, FOXO3, has been found in another study to contribute to muscle wasting by activating both autophagy and proteasomal degradation. 91 This study showed that FOXO3 promoted different degrees of activation of the two processes in different cell types. Specifically, both autophagy and proteasomal degradation were promoted at the same degree in muscle fiber cells while only autophagy was activated down-stream of FOXO3 in hepatoma and pheochromocytoma cells. 91 Expression of a plasmid encoding a dominant-negative FOXO in mouse muscle cells abrogated wasting. 92 In the low nutrient environment of muscle in cancer, FOXO3 coordinates a stress response associated with a catabolic state. 93 These results support a role for the FOXO3 isoform in cancer-associated muscle wasting through both autophagic and proteasomal protein degradation. Dysregulation of insulin and insulin-like growth factor 1 (IGF-1) signaling axis that inhibits FOXOs is involved in cancer-associated wasting. 94 In sympathetic neurons, FOXO3 is activated after nerve growth factor (NGF) withdrawal and co-operates with NF-Y on the bim promoter to induce this pro-apoptotic member of the bcl-2 family. 95 This may be a more general theme relevant for proteasome regulation in cancer, whence activated cell surface growth factor cascades inhibit FOXOs through Akt activation and would impair the co-operation of FOXOs with NF-Y in proteasome component genes promoters (Figure 8). Suppressed proteasome activity in cancer stem and tumor-initiated cells may be explained by FOXO activity suppression, while additional regulations, for example, through NRF1/NRF2 activation down-stream of Akt, may override this suppression in bulk cancer cells.
A third FOXO family member, FOXO1, is involved in the protection of leukemic cells against toxicity induced by pharmacologic proteasome inhibition. 96 FOXO1 was induced when leukemic cells were exposed to the polyphenol resveratrol, a dietary compound found in grapes, and this induction protected cells from the apoptotic effects of proteasome inhibitors MG-132 and lactacystin. In addition, FOXO1 induced cell cycle arrest through transcriptional up-regulation of cyclin kinase inhibitor p27Kip1. 96 Proteasome sub-unit induction was not specifically checked in this study but could be a more direct additional mechanism through which FOXO1 induction protects from proteasome inhibition in these cells. In this respect, another study reported that repression of PSMB5, PSMB10, and PA28α sub-units sensitized hepatocellular carcinoma cells to bortezomib. 97
STAT3 is a transcription factor activated down-stream of several receptor tyrosine kinases, including EGFR, and cytokine receptors (Figure 7). It is activated in several cancer cell types and has been shown to up-regulate several β sub-units of the 20S proteasome. 98 Prostate cancer cells with activated STAT3 at baseline displayed a decrease of proteasome sub-unit PSMB5 mRNA and protein levels when STAT3 was dephosphorylated by inhibitors Stattic and WP1066, and thus, inactivated. Such response was not observed in another prostate cancer cell line without baseline STAT3 activation. 98 Thus, in certain cancers, STAT3 pathway activation may contribute to proteasome up-regulation down-stream of growth factors. Of interest, STAT3 is activated in CSCs, 99 and this implies that additional signals suppress the expression of proteasome sub-units in these cells, which become de-repressed in the differentiated progeny, or alternatively, additional activating signals up-regulate proteasome units in these progeny cells. 19S sub-units or α ring units of the 20S proteasome and their regulation by STAT3 were not specifically investigated in the above study 98 or any other studies, but multiple STAT3 binding sites are present in all components promoters as checked in the Transcriptional Regulatory Element Database (TRED) and JASPAR databases (see later). STAT3 is also involved in cancer cachexia by stimulating proteolysis through activation of both the proteasome and apoptosis-related protease caspase 3 in muscle of cancer-bearing animals. 100
In contrast to the proteasome function that is decreased in CSCs, the expression of individual sub-units may be increased. This is the case for sub-unit PSMD10 (Gankyrin) that shows an increased expression in colon CSCs, and its expression correlates with the expression of the stem cell marker CD133. 101 Enforced expression of PSMD10 in hepatoma cells promoted dedifferentiation and also increased CD133 expression. 102 Regulation of PSMD10 transcription is activated by β-catenin and c-myc, 103 in addition to the general transcription panel members regulating other proteasome units expression, such as NRF2. 104 PSMD10 is a ubiquitin receptor and is involved in facilitation of degradation of both tumor suppressors p53 and Rb by the proteasome. 105 Moreover, in hepatocellular carcinoma cells, PSMD10 interfered with the interaction of Oct4 with E3 ligase WWP2, impeding the degradation of the pluripotency transcription factor and promoting the expansion of tumor-initiating cells. 106 PSMD10 promoted, additionally, degradation of differentiation transcription factor HNF4α (hepatocyte nuclear factor 4α). 102 Thus, PSMD10 up-regulation in CSCs may lead to preferential use of low residual proteasome activity present in these cells toward tumor suppressors and differentiation factors and away from pluripotency inducers.
A common property of all transcription factors regulating proteasome is that they are substrates of the proteasome degradation activity and thus participate in negative feedback loops. 107 These loops are deregulated if exogenous factors such as oncogene activation or pharmacologic proteasome inhibition interfere with activity of a part of the loop. Feedback deregulation is particularly relevant in cancer where there is a universal abnormal activation of growth factor pathways. Signals from these pathways appear to co-operate with feedback signals from increased proteostasis requirements in order to up-regulate proteasome activity in bulk cancer cells but are not sufficient to up-regulate this activity in CSCs where the proteostasis needs are lower as these cells are generally more quiescent. Pharmacologic inhibition of the proteasome itself by MG-132 or lactacystin up-regulates the activity of transcription factors HSF1 and HSF2 and increases levels of targets heat shock protein 72 (HSP72) and interleukin 6 (IL-6). 108 HSF2 up-regulation and IL-6 up-regulation, through STAT3, may additionally result in proteasome activity feedback up-regulation by production of new sub-units, if proteasome inhibitors exposure persists.
In contrast to the negative regulation of transcription factors by proteasome degradation, a positive regulation of NRF2 by the proteasome component protein PSMD10 (Gankyrin) has been observed. 104 In this case, PSMD10 competes with NRF2 for Keap1 which subsequently leads to proteasome degradation. Rescue of NRF2 by this mechanism that was discovered in hepatocellular carcinoma cells results in production of new PSMD10 and other proteasome units that are transcriptional targets of NRF2 in a positive feed-forward loop. However, a proteomic analysis in colorectal cancer cells showed that knock-out of PSMD10 resulted in down-regulation of other proteasome units, including PSMB4, PSMB7, and PSME2. 109 In cholangiocarcinoma, PSMD10 was reported to increase activity of STAT3 with a mechanism involving promotion of Rb phosphorylation and increasing IL-6 signaling. 110
Several proteasome sub-unit promoters exhibit binding sequences of the transcription factors that have been experimentally implicated in proteasome regulation and discussed above. An evaluation of all promoters of proteasome sub-units and assembling proteins listed in the TRED (https://cb.utdallas.edu/cgi-bin/TRED/tred.cgi?process=home) through the JASPAR transcription factor binding profile database (http://jaspar.genereg.net/; Figure 3) revealed that almost all of these promoters possess multiple FOXO and STAT3 binding sites. In addition, several of the component genes promoters possess binding sites for the three other transcription factors NF-Y, NFE2L1 and 2, and HSF2. Tables 1 and 2 present the number of putative binding sites of each transcription factor in each promoter of proteasome sub-units of the β ring of the 20S proteasome and of the base of the 19S proteasome, as listed in TRED and checked into JASPAR. No obvious unifying theme of “en bloc” regulation is evident. Nevertheless, given that, experimentally, changes in levels of individual sub-units have been shown to change proteasome activity, and that all transcription factors discussed are part of feedback loop regulations by the proteasome, a coordinate system network may still exist. This would be facilitated by the fact that the stoichiometry of units is not rigid. As discussed above, proteasomes with more than two CP sub-units of one kind and less than two of another exist, and, similarly, RP units may be replaced by alternative regulators.
Number of transcription factor binding sites in promoters of component genes of the β ring of 20S proteasome.

TRED: Transcriptional Regulatory Element Database.
All the promoters listed in the TRED database were checked using the JASPAR database of binding sites. Logos of transcription factor binding sites are depicted in Figure 3.
Number of transcription factor binding sites in promoters of component genes of the base of 19S proteasome.

TRED: Transcriptional Regulatory Element Database.
All the promoters listed in the TRED database were fed into the JASPAR database of binding sites. Logos of transcription factor binding sites are depicted in Figure 3.
It is appropriate to note that investigation of promoter binding sites is in general disfavored when transcription factors bind degenerate sites that significantly deviate from their canonical site, and this appears to be the case, for example, for NF-Y, due to the need to accommodate co-operative transcription factors binding. 111 As a result, the current in silico investigation, despite providing information on the landscape of proteasome sub-unit promoters, may be of limited value for the determination of actual functional regulatory interactions.
In addition to transcriptional control of proteasome sub-units, various post-translational regulations take place that affect protein expression of the sub-units (see also next section for a brief discussion). Thus, mRNA expressions may not always correlate with protein levels in various tissues and cancers. In fact, a survey of the Human Protein Atlas (www.proteinatlas.org) for levels of proteasome units disclosed that, compared to normal tissues of the respective organs, three common cancers, breast, colorectal, and lung, had a lower expression of most proteasome sub-units at the protein level when investigated by immunohistochemistry (data not shown).
Post-translational regulation of proteasome units
In addition to regulation of the proteasome at the level of expression of its component genes, sub-unit proteins are regulated in the post-transcriptional level. For example, PSMD11 directly interacts with kinase AMPK and may be phosphorylated in two consensus kinase sequences that are present in the protein. 112 Phosphorylation may lead to subsequent ubiquitination of PSMD11 and its own degradation. In response to genotoxic stress after exposure to doxorubicin, several units of the 20S CP are phosphorylated and ubiquitinated. 113 Ubiquitination of sub-unit Rpn13 (gene PSMD4) takes place when proteasome proteolytic function is inhibited and prevents further ubiquitinated proteins to bind the non-functioning particle. 114 SUMOylation of unit PSMD1 is required for the interaction of Rpn13 with the proteasome. 115 Phosphorylation is also required for facilitation of sub-units incorporation during proteasome assembly. 116 Post-transcriptional regulations of proteasome sub-units are of equal importance with transcriptional regulation of their production for proteasome abundance and function. For an overview of the life cycle of proteasome that includes a more detailed discussion of post-transcriptional regulations, interested readers are referred to a recent review. 117
Therapeutic perspectives
Regulation of the multiple proteasome units at the transcriptional level is carried out by multiple transcription factors which in addition have multiple partners and are subjects to various regulations in their turn. This multitude of networks makes the coordinated theme of regulation difficult to decipher. An additional difficulty results from the fact that most investigations of molecular mechanisms are carried in unsorted cancer cells and cell lines and thus do not depict the tumor stem cell compartment. Nevertheless, identification of decreased proteasome activity in CSCs in association with up-regulation of individual units such as PSMD10 provides a framework for formulating a hypothesis of how this decreased activity is embedded in the pluripotency of CSCs in order to inform possible therapeutic interventions. The picture arising in CSCs points to the low residual proteasome activity being biased toward the destruction of specific target proteins such as tumor suppressors p53 and Rb and sparing others such as the pluripotency factor Oct4. Similar to development, changes in proteasome activity are observed when CSCs differentiate to non-cancer stem cell counterparts, while a level of plasticity is maintained and proteasome function inhibition may help non-stem cells to obtain cancer-initiating properties. 35 This would also explain why pharmacologic proteasome inhibition has not produced the hoped-for results in cancer therapeutics, besides multiple myeloma and sub-types of lymphoma which may be particularly sensitive due to proteostasis requirements related to antibody handling.118,119 A more specific targeting of proteasome machinery components may be a more effective alternative to inhibition of enzymatic β sub-units. The obvious choice based on the above discussion would be inhibition of PSMD10 (Gankyrin). Small molecule inhibitors and synthetic proteins inhibiting Gankyrin and restoring p53 levels in cells have been discovered and are possible candidates for further development.120,121 A small molecule inhibitor called cjoc42 was shown to bind to the protein–protein interaction surface of PSMD10, preventing its interaction with 19S proteasome component ATPase S6 (PSMC4). 120 This blocked interaction leads to stabilization of wild-type p53 protein in osteosarcoma cells overexpressing PSMD10, when these cells are treated with cjoc42, probably because of interference with the delivery of p53 to proteasome for degradation. A synthetic artificial protein is an alternative for blockade of the large PSMD10 protein–protein interaction surface, if it can be successfully delivered to cells in vivo. A scaffold synthetic protein called GBP7.19 was designed in silico and then optimized to tightly and selectively bind PSMD10 with a KD of about 21 nM. 121 GBP7.19 blocks the interaction of PSMD10 with a hybrid protein containing the carboxyterminal part of PSMC4 in yeast cells and the ubiquitination of p53 in an in vitro assay. These results confirm the feasibility of PSMD10 blockade in cells and the effect that this blockade has in increasing p53 stability and constitute the basis for further pre-clinical and eventual clinical development for cancers with a wild-type p53 protein.
Another therapeutic opportunity could present at the level of proteasome substrate de-ubiquitination. PSMD14 is the intrinsic proteasome de-ubiquitinase sub-unit that dissociates the ubiquitin chain from the target proteins after the latter have been committed to degradation. In contrast to PSMD14, proteasome-associated de-ubiquitinases USP14 and UCH37 cut ubiquitins from the periphery of the chain one at a time interfering with, instead of facilitating, degradation. As a result, a small molecule inhibitor of USP14 has been found to increase proteasome activity. 122 This could be exploited to increase proteasome activity and lead to differentiation of CSCs, although this remains to be proven experimentally in vivo.
A genomic investigation of cancer cell lines aiming at discovering genes which produce cancer vulnerabilities when haploinsufficient (called CYCLOPS genes for copy number alterations yielding cancer liabilities owing to partial loss) disclosed that proteasome components PSMC2 and PSMA4 were among these genes together with components of the spliceosome and the ribosome. 123 When PSMC2 was knocked-down with RNA interference (RNAi), haploinsufficient cells showed proliferation inhibition and increased apoptosis compared with diploid cells. The proposed mechanism of vulnerability is that higher amount of PSMC2 in diploid cells creates a reservoir of the protein that may be used for 26S proteasome insertion or other cellular functions of the 19S proteasome when new supply is knocked-down, while haploinsufficient cells do not have this extra cushion. 123 These data argue for a role of individual proteasome sub-units inhibition in cancers with specific molecular lesions.
An additional benefit of inhibiting 19S proteasome components without affecting the CP is that this inhibition may favor association of alternative activators, such as the 11S unit, which promotes production of peptides presentable to the immune system. This could have implications for the immunogenicity of tumor cells and CSCs. Immune presentation of antigens has become even more important in cancer therapeutics recently with the introduction of immune blockade inhibitors in the clinic. 124
A more in-depth understanding of the proteasome regulation in the individual sub-unit level in cancer in general and in CSCs in particular will certainly unveil further opportunities in cancer therapeutics and disclose individual cancer sub-types more amenable to proteasome manipulation for therapeutic benefit. Nevertheless, low proteasome activity as an inbuilt characteristic of CSCs may facilitate a more universal targeting with proteasome inhibitors, possibly in combination with specific inhibitors of pathways important in individual cancers.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.