
Abstract
Background/Purpose
Alzheimer’s disease (AD) is a long-term neurodegenerative condition that leads to the gradual deterioration of nerve cells. The goal of this study is to use computational drug discovery (CDD) techniques to discover lead compounds that target Amyloid-beta A4 (AβA4) as a potential target for AD.
Materials and Methods
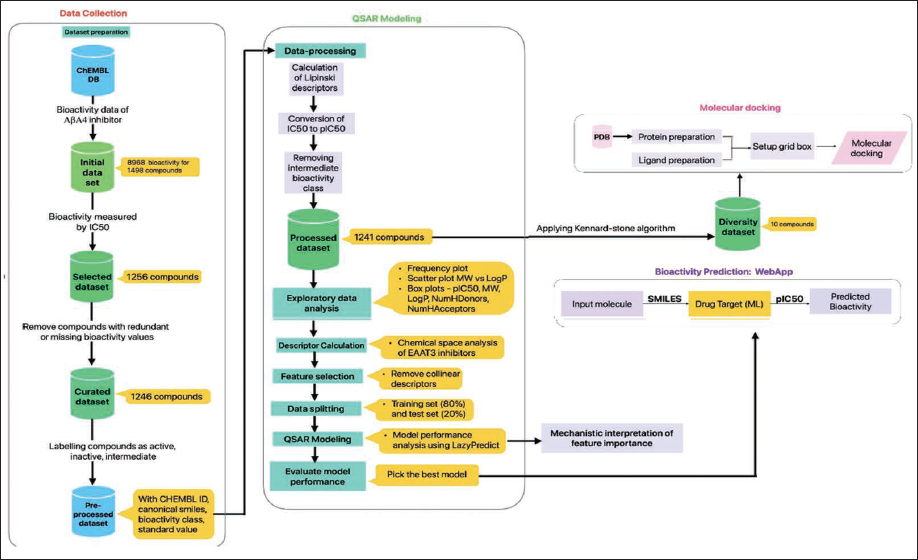
Quantitative structure-activity relationship (QSAR) modeling is used in this study to compare different machine learning (ML) models aimed at predicting the potency negative logarithm of IC50 (pIC50) of candidate compounds, which are then validated by molecular docking based on their binding affinity. A non-redundant dataset consisting of 1,241 compounds for AβA4 was retrieved from the ChEMBL database. 880 substructure fingerprints were used to define these compounds, followed by building 42 ML models and comparison. The Kennard–Stone algorithm was employed to select a diverse set of 30 compounds from the set of active inhibitors for testing. The application programming interface (API), named NeuroIC50, was developed and deployed.
Results
The histogram-based gradient boosting regression (HGBR) tree has achieved the optimal performance compared to other regression models, as determined by its root mean square error (RMSE) of 0.73, R2 value of 0.65, and time efficiency of 0.78. Random forest regression (RFR)-HGBR-derived Gini index revealed the importance of features, include SubFP23, SubFP405, and SubFP577 in the compounds.
Conclusion
The lead compound (CHEMBL5080033) with a pIC50 of 8.67 M and a binding energy of −7.6 kcal/mol was identified. This ML-based QSAR modeling and docking approach is an effective strategy for accelerating drug discovery.
Keywords
Introduction
Alzheimer’s disease (AD) is a slowly progressing neurodegenerative disorder, resulting in memory problems, reduced cognitive ability, and difficulty in carrying out daily activities. 1 It is caused by the build-up of amyloid plaques and neurofibrillary tangles, which leads to the death of brain cells and synaptic dysfunction. 2 AD progresses through stages from mild to severe, with symptoms worsening over time. 3 Global estimates of AD and other forms of dementia suggest that the burden on society is escalating at a concerning pace. In 2006, the worldwide prevalence was 26.6 million, projected to quadruple by 2050, 4 and more recent data suggest even higher estimates, with 51.6 million affected in 2019 5 and 44.35 million in 2013, expected to reach 135.46 million by 2050. 6 Interventions delaying disease onset and progression by just 1 year could significantly reduce the global burden. 7 However, there are not many effective drugs for the treatment of AD. Despite the high prevalence, the symptoms associated with AD, and the significant unmet medical need, many pharmaceutical companies have moved away from drug discovery, development, and research for AD spectrum disorders, due to several factors, including high costs, high failure rates in clinical trials, a poor understanding of the disease’s underlying mechanisms, and the lengthy time required for drug development. 8
Amyloid-β (Aβ) plaque accumulation represents a pathological characteristic of AD, occurring 15–20 years prior to the onset of clinical symptoms. These plaques trigger a series of biological events, including neuroinflammation, synaptic dysfunction, along with challenges in tau pathology, oxidative stress, protein clearance, mitochondrial function, and calcium homeostasis. 9 The amyloidogenic pathway produces Aβ peptides, notably Aβ40 and Aβ42, with the accumulation and aggregation of Aβ being associated with AD. 10 Targeting Aβ remains a promising therapeutic approach, despite recent setbacks. Existing AβA4 inhibitors include compounds like Lecanemab, Aducanumab, Valiltramiprosate, Donanemab, and others. 11
Accurately predicting the activity of modulators of Amyloid-beta A4 (AβA4) is a complex task because of the diverse nature of transporters and their interactions with inhibitors. To address this complexity, the incorporation of machine learning (ML) techniques into quantitative structure-activity relationship (QSAR) has proven to be a valuable tool. QSAR plays a pivotal role in establishing relationships between chemical structures and biological activity, using statistical or ML models. 12
ML is a branch of artificial intelligence (AI) that focuses on the development and usage of computer algorithms that are capable of learning from unprocessed data in order to perform designated tasks effectively. 13 AI algorithms engage in tasks such as classification, regression, clustering, and so on, across extensive datasets. In the pharmaceutical realm, a diverse array of ML techniques has been used to predict novel molecular descriptors, biological functions, activities, interactions, and adverse effects of drugs. 12 Examples of regression models include decision trees, random forests, linear regression, and neural networks. 14 In contrast, popular classification models include logistic regression, support vector machine, and naive Bayes. 15 However, the Lazy Predict library in Python, 16 can be employed to find the best model for classification and regression based on our data, without any parameter tuning. LazyClassifier can be used to classify the datasets into an 80% training set and a 20% test set, while LazyRegressor can be used to compare the performances between several regression models. 17
We present a comprehensive QSAR pipeline aimed at predicting AβA4 inhibition using 1,241 non-redundant compounds. Interpretable learning methods like random forest and molecular descriptors, such as molecular fingerprints, were applied to uncover the inhibitory activity of AβA4 in accordance with Organization for Economic Co-operation and Development (OECD) guidelines. Additionally, molecular docking studies were conducted on selected active AβA4 inhibitors. This combined ligand- and structure-based approach aims to provide insights into the design of selective AβA4 inhibitors, offering a novel therapeutic strategy for AD. To support this, we developed NeuroIC50, a computational server that predicts the inhibition potency, negative logarithm of IC50 (pIC50) of ligands using a QSAR model. Researchers can input ligands as SMILES, screen inhibitory potential, and validate efficacy through pIC50 values. This tool aims to advance the identification of potent AβA4 inhibitors and improve AD treatment strategies.
Materials and Methods
Dataset
A dataset of inhibitors targeting human AβA4 (Target ID CHEMBL2487) was compiled from the ChEMBL database, comprising a total of 8,968 bioactivity data points derived from 1,498 compounds. We refined the SMILES notations of the compounds using ChemAxon’s standardizer, adhering to the parameters set forth in the study by Simeon. 18 Initially, we built the dataset from various bioactivity measurement units, sorted by decreasing data size: IC50, Ki, %activity, %inhibition, EC50, and so forth. We chose to focus on the IC50 values for further investigation, as they represented the largest subset with 1,256 compounds. Upon closer examination, we identified nine compounds lacking reported IC50 and canonical SMILES. Henceforth, they were excluded from the study, thereby leaving 1,247 compounds. Additionally, we retained redundant compounds with differing bioactivity values only if the standard deviation of IC50 values was less than two, and this resulted in a refined dataset of 1,243 compounds. We converted the IC50 values into pIC50 values, using the relation pIC50 = −log10(IC50), so that we have normalized data that are suitable for the ML pipeline. We categorized compounds with pIC50 values >5 as active, <5 as inactive, and = 5 as intermediate. We removed the intermediate class, leading to a final dataset of 1,241 compounds. A summary of this pipeline is illustrated in Figure 1.
Workflow of Quantitative Structure-activity Relationship (QSAR) Modeling and Molecular Docking for Investigating an Inhibitory Activity.
Molecular Descriptors of Inhibitors
Molecular descriptors are quantitative or qualitative representations that characterize molecules based on their structure, connectivity, and physicochemical characteristics. They play a crucial role in QSAR studies. These descriptors can be computed using graphical user interface (GUI)-based software like Dragon, PaDEL-Descriptor, QuBiLS-MIDAS, QuBiLS-MAS, and CODESSA. Alternatively, they can be computed using R or Python libraries, such as ChemoPy, PyDPI, RDKit, and rcdk. Additionally, web servers like BioTriangle and ChemDes also offer molecular descriptor calculation services online.19–21 Fingerprint descriptors capture information about the underlying substructures inherently present in a molecule. In this study, we utilized RDKit for computing 881 PubChem fingerprints. 22 Additionally, we computed four molecular descriptors that define Lipinski’s rule of five, comprising molecular weight (MW), logarithm of octanol/water partition coefficient (ALogP), number of hydrogen bond donors (nHBDon), and number of hydrogen bond acceptors (nHBAcc), using RDKit.
Feature Selection and Data Splitting
Collinearity refers to a condition where descriptor pairs exhibit intercorrelation, which results in model complexity and potential bias. To address this issue, we employed the corr( ) function from the Python library, pandas, to calculate the pairwise correlation among the descriptors. We excluded the descriptors with a Pearson’s correlation coefficient greater than the threshold of .7 for further study. 23 Following this, we splitted the dataset into two sets, namely, the training set and the test set, with the former comprising 80% and the latter comprising 20% of the original dataset. We utilized the train_test_split( ) function from the sklearn.model selection Python library to facilitate the data split.
Multivariate Analysis
Supervised learning is training a model from labeled data that can be used to make predictions about unseen or future data. 24 In this study, the Python library, LazyPredict, was used to construct multiple regression models and evaluate their performances, and these models aim to predict the continuous response variable (pIC50) using predictor variables (fingerprint descriptors). This study primarily employs the histogram gradient boosting regressor (HGBR). HGBR represents an ensemble method grounded in gradient boosting principles, wherein decision trees are built sequentially, where each tree attempts to rectify the inaccuracies made by its predecessors. It integrates histogram-based techniques for accelerated training on extensive datasets. 25 The HistGradientBoostingRegressor() function from the Python library scikit-learn was used to construct the models, providing an efficient implementation of the HGBR algorithm. 26 For a better understanding of the key substructures of the compounds responsible for the modulatory activity, we extracted informative descriptors from the HGBR model using the built-in feature importance estimator. Specifically, the mean decrease of the Gini index (MDGI) was employed to identify the most important descriptors (descriptors with the highest MDGI values).
Validation of QSAR Models
Model validation plays a crucial role to confirm that a fitted model can reliably predict responses for future or unseen subjects. In this study, we evaluated the performance of the QSAR models using three statistical parameters, namely, R2, root mean square error (RMSE), and computation time. R2 quantifies the proportion of variance in the dependent variable explained by the independent variables. RMSE assesses the average magnitude of the errors between predicted values and actual values. Computation time refers to the time required to train the model and make predictions. While time is an important consideration in real-world applications, it does not directly affect model accuracy. A shorter time is preferred, especially for real-time applications or when dealing with large datasets. In summary, models with higher R2, lower RMSE, and computation time are preferred indicators of quality.
Server Deployment
We have developed and successfully deployed NeuroIC50, a web application designed using the Streamlit (50), framework for predicting the bioactivity of chemical compounds targeting AβA4, a crucial target in AD research. The application offers a comprehensive, interactive interface that seamlessly integrates various functionalities, such as molecular descriptor calculation and ML-based prediction. Users can easily upload their molecular data in text format, after which the application computes molecular descriptors utilizing the PaDEL-Descriptor tool, executed via a subprocess, employing the same PubChem fingerprint calculation protocol used during model development. These calculated descriptors serve as input for a pre-trained ML model, which then predicts bioactivity in terms of pIC50 values. The results are dynamically displayed on the app and can be downloaded in comma-separated values (CSV) format, ensuring an efficient and user-friendly experience for bioactivity prediction tasks.
Results
Chemical Space of AβA4 Inhibitors
We carried out a chemical space analysis of AβA4 inhibitors to gain insights into the structure-activity relationship (SAR) by examining Lipinski’s rule of five descriptors, consisting of MW, the logarithm of the partition coefficient between n-octanol and water (ALogP), the nHBDon, and the nHBAcc. MW, a key size indicator, plays a key role in a compound’s ability to pass through lipid membranes, while ALogP, a commonly used indicator of a compound’s lipophilicity, is important for determining membrane permeability. nHBDon and nHBAcc describe the hydrogen bonding potential of a compound, relevant for assessing hydrogen bond formation capability.
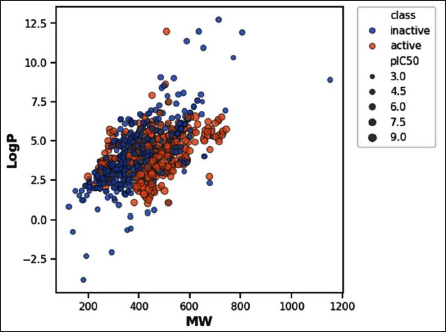
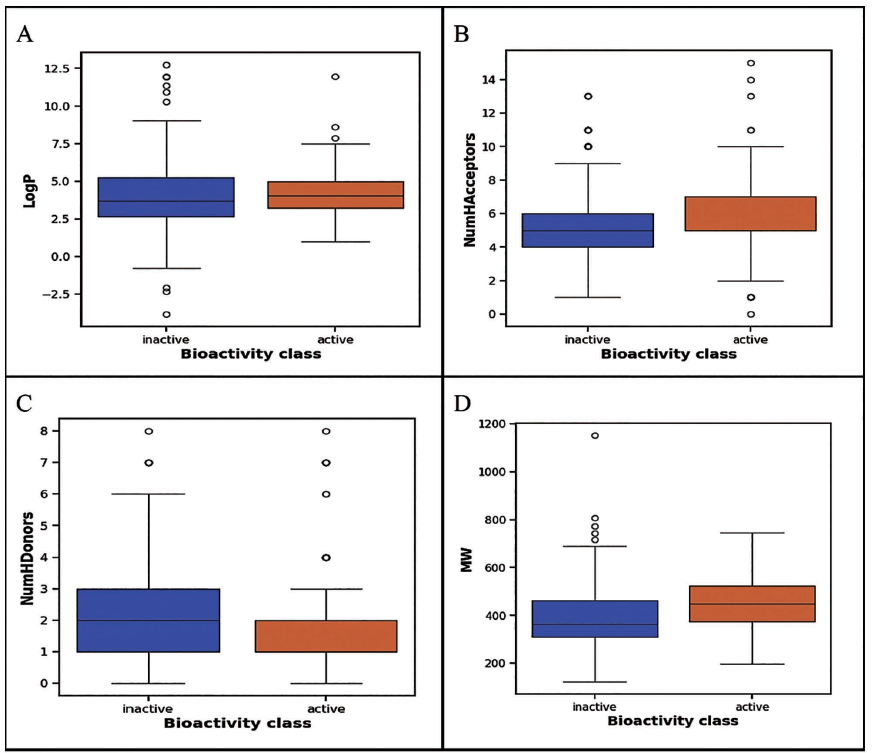
Visualization of the chemical space of AβA4 inhibitors, particularly ALogP as a function of MW (Figure 2), revealed a dense clustering of inhibitors in the MW range of 200–600 Da and ALogP values between 2 and 7. ALogP, nHBAcc, nHBDon, and MW are shown in Figure 3A–3D, respectively. The Mann–Whitney U test was used to statistically compare the descriptors between active and inactive compounds. The analysis demonstrates that there are significant differences in the nHBDon and nHBAcc. Specifically, active compounds exhibit lower values of nHBDon and nHBAcc compared to inactive compounds. In contrast, while there were observable differences in LogP and MW, these were not statistically significant based on the Mann–Whitney U test.
Chemical Space of Amyloid-beta A4 (AβA4) Inhibitors. Active and Inactive are Shown in Red and Blue, Respectively.
Boxplot of Amyloid-beta A4 (AβA4) Inhibitors Using Lipinski’s Rule of Five Descriptors. (A) Box Plots of ALogP, (B) Box Plots of Number of Hydrogen Bond Acceptors (NumHAcceptors), (C) Box Plots of Num Number of Hydrogen Bond Donors (NumHDonors), and (D) Box Plots of Molecular Weight.
QSAR Model for Predicting AβA4 Inhibitory Activity
We utilized the dataset consisting of 1,241 compounds to construct QSAR models. We generated molecular descriptors for this dataset using the PaDEL-Descriptor module in Python, which offers a wide range of fingerprint descriptors. For this study, we explicitly employed 881 PubChem fingerprint descriptors for model development and benchmarking. Later, we performed feature selection to eliminate collinear descriptors, and 42 models were built using an 80/20 train-test split. Performance results are summarized in Table 1, with the best models characterized by high R2 values, low RMSE, and minimal overfitting.
Performance Summary of Training Set for Predicting Negative Logarithm of IC50 (pIC50).
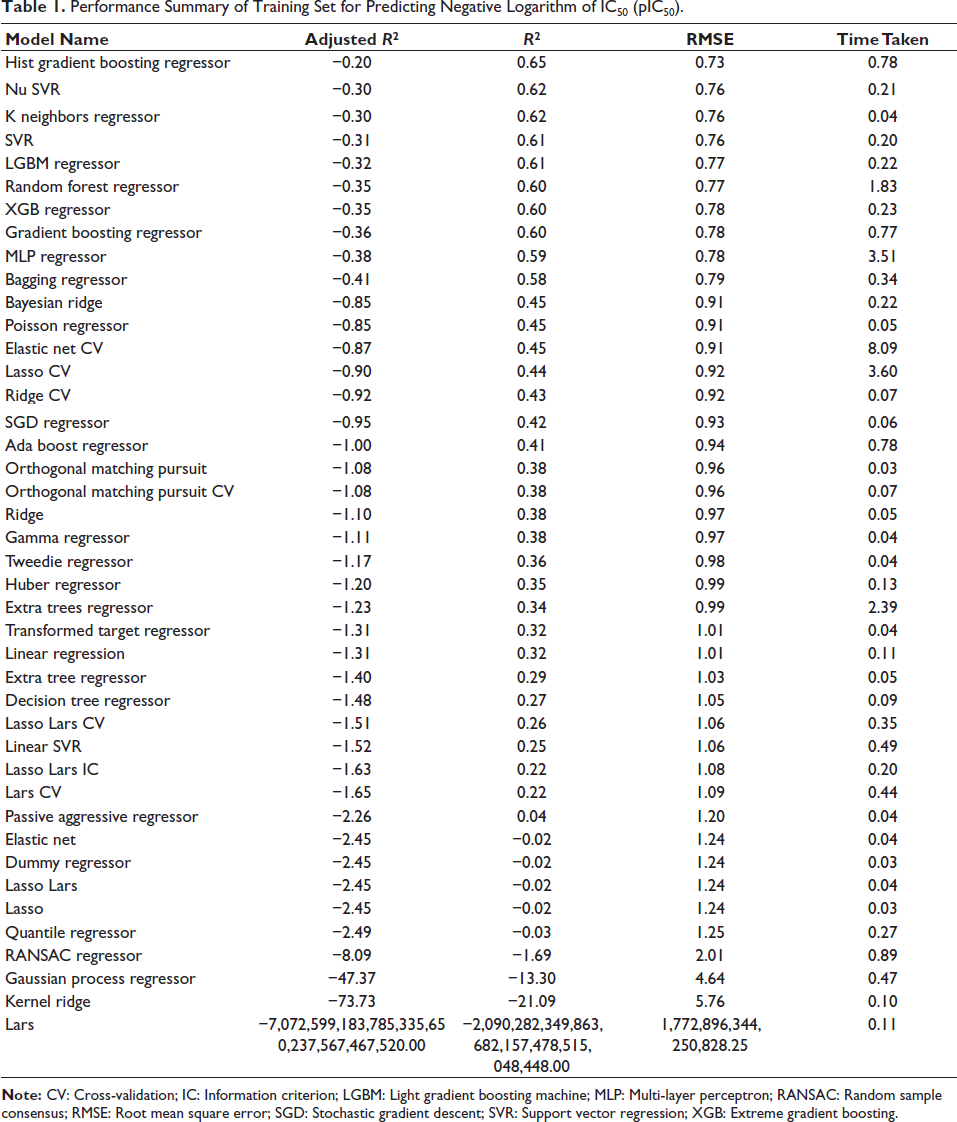
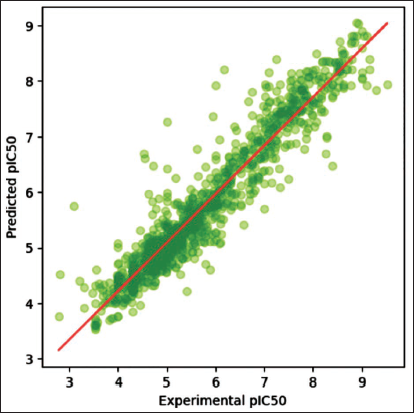
Histogram gradient regressor achieved the highest training set R2 (0.65) and a low RMSE (0.73), while the nu support vector regression yielded an R2 of 0.62 and an RMSE of 0.76, as shown in Table 1. Upon evaluation on the test set, these two models exhibited consistent performance across both sets, suggesting they learned patterns applicable to new data, making them less prone to overfitting. Although it showed a negative adjusted R2 value, that is, −0.20, this does not arise due to poor performance but rather due to the metric penalizing additional features that do not contribute sufficient explanatory gain. As illustrated in the scatterplot (Figure 4), predicted pIC50 values are plotted against experimental pIC50 values. Most data points cluster around the diagonal line, indicating that the HGBR effectively captures the relationship between molecular descriptors and pIC50 values. This positive correlation signifies the model’s accuracy across the dataset.
Plot of Experimental Versus Predicted Negative Logarithm of IC50 (pIC50) Values for Models Constructed with 881 PubChem Fingerprint Descriptors.
Mechanistic Interpretation of Feature Importance
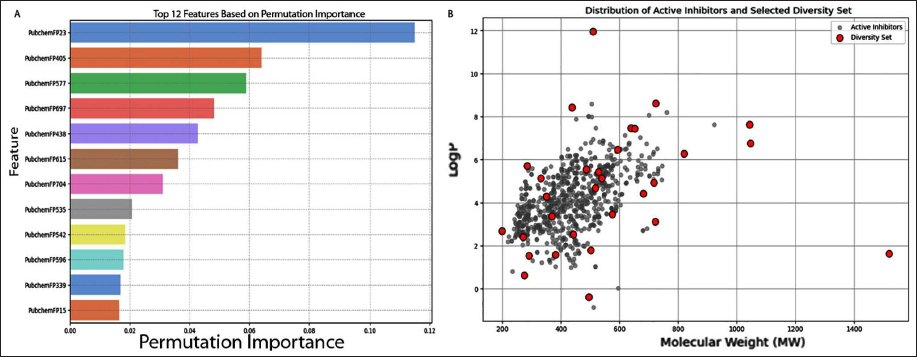
Feature importance analysis helps identify key features that contribute towards bioactivity. When evaluating the relative importance of features in models using the HGBR algorithm, two parameters mainly come into play: (a) SHapley Additive exPlanations (SHAP) and (b) Gini index (i.e., variance of the responses). The latter was selected as a metric for ranking important features (i.e., the MDGI) for predicting the pIC50 of AβA4 inhibitors (Figure 5A). Table 2 lists the substructure fingerprints, along with their respective descriptions.
(A) Plot of Mechanistic Feature Importance as Exemplified by the Gini Index. (B) Plot Showing the Distribution of Active Amyloid-beta A4 (AβA4) Inhibitors (Gray Circles) and the Diversity Set (Red Circles) Selected for Molecular Docking.
List of Top PubChem Substructure Fingerprints and Their Corresponding Description.

In Figure 5A, we can observe that the top-ranking feature is >=1F (PubChemFP23), which indicates the presence of at least one fluorine atom. In the context of drug design, its small size and electronic properties allow it to influence drug-receptor interactions through hydrophobic, electrostatic, and hydrogen-bond interactions, often leading to enhanced drug activity. 27 The second most important feature is O(~C) (~C) (PubChemFP405); it refers to an oxygen atom single-bonded to two carbon atoms. Oxygen atoms in various functional groups, including ethers and esters, exhibit distinct electron density configurations that influence hydrogen bonding directionality. Allylic structures containing oxygen atoms can enhance drug potency and toxicity. These unconventional hydrogen bonds, involving oxygen as an acceptor, contribute significantly to ligand-receptor interactions and are crucial in drug discovery. 28
The third most important feature is C:C-N-C:C; a nitrogen atom single-bonded to two carbon atoms, which resembles the pattern of enamines (PubChemFP577). These are central to biological catalysts like aldolases and small-molecule amine organocatalysts. 29 The fourth most important feature is C-C-C-C-C-C(C)-C, a branched alkane, 2-methylhexane. Introducing methyl groups into small molecules has become an important strategy for lead compound optimization, improving metabolic stability and generating new uses for existing drugs (PubChemFP697). 30 The fifth most important feature is C(-C)(-N)(=N); resembling guanidine (PubChemFP438). Tryptophan-derived guanidine compounds, such as TGN2, have shown potential as multifunctional agents, inhibiting both BACE1 and amyloid aggregation while demonstrating neuroprotective effects. 31 The sixth most important feature is N-C-C-O-C, resembling amino alcohols or amino ethers (PubChemFP615). Tailor-made amino acids, including their modifications like diamines and amino alcohols, are key structural features in many blockbuster drugs, demonstrating their importance across various therapeutic areas. 32 The seventh, eighth, ninth, and tenth important features are O=C-C-C-C-C-C-C; heptanoic acid/enanthic acid (PubChemFP704), O=C-C-C; propanoic acid (PubChemFP535), O-C:C-[#1]; corresponding to the enol ether functional group (PubChemFP542), and N=C-N-C-C; representing a characteristic of substituted guanidine derivatives (PubChemFP596), respectively. 33
Discussion
Our study evaluates a diverse set of chemical compounds for their ability to inhibit AβA4, a therapeutic target in AD. Using a combination of ML-based QSAR modeling and molecular docking, we evaluated the bioactivity of candidate compounds (Figure 5B). Our study shows that CHEMBL5080033 has a predicted pIC50 of 8.67 and a binding energy of −7.6 kcal/mol, making it a promising lead for future therapeutic development.
The chemical space analysis of AβA4 inhibitors highlighted that active compounds tend to have lower numbers of hydrogen bond donors and acceptors, suggesting that steric and hydrogen-bonding features influence binding interactions. We also observed that the molecular descriptors, such as MW and lipophilicity, cluster within ranges of favorable pharmacokinetic properties, further supporting their potential as drug-like candidates.
And, HGBR outperformed the other 41 models in predicting pIC50 values, as shown by its R2 of 0.65 and an RMSE of 0.73. We identified key substructural fingerprints contributing to inhibitory activity via the HGBR-derived Gini index, which includes fluorine atoms (PubChemFP23), oxygen ethers/esters (PubChemFP405), enamines (PubChemFP577), branched alkanes (PubChemFP697), guanidine motifs (PubChemFP438), and amino alcohols/ethers (PubChemFP615). These functional groups enhance ligand-receptor interactions through a combination of hydrogen bonding, electrostatic, and hydrophobic interactions, which provide insights into SARs and help in lead optimization.
Molecular docking studies further support our QSAR findings, showing stable interactions between selected inhibitors and the AβA4 target through hydrogen bonds and hydrophobic contacts. These results suggest that the identified lead compounds not only possess favorable predicted bioactivity but also form energetically stable complexes with their target.
While the current study provides strong computational evidence for promising AβA4 inhibitors, further experimental validation is necessary to confirm biological activity. Additionally, molecular dynamics simulations could be employed in future work to assess the stability and conformational flexibility of ligand-bound complexes, which could help in understanding the mechanistic basis of inhibition and supporting the advancement of these compounds toward clinical viability.
Conclusion
In conclusion, we utilized 881 PubChem fingerprint descriptors for developing QSAR models, and we evaluated their comparative performances. We observed that Histogram Gradient Regressor demonstrated strong performance for the constructed models, indicating that they could capture the feature space of AβA4 inhibitors. Utilizing the Gini index, a built-in feature importance estimator of the HGBR, we were able to identify key features essential for AβA4 inhibition: PubChemFP23, PubChemFP405, PubChemFP577, PubChemFP697, PubChemFP438, PubChemFP615, PubChemFP704, PubChemFP535, PubChemFP542, and PubChemFP596. The insights gained from this research are expected to serve as general guidelines for the development of novel AβA4 inhibitors.
Footnotes
Abbreviations
Authors Contribution
SRK designed and performed the experiments. SRK and IMS analyzed the data. SRK wrote the original draft, and SRK and IMS contributed to the review and editing of the manuscript. SPD conceptualized, reviewed, and edited the manuscript and supervised the study.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval was obtained from the relevant ethics committee or Institutional Review Board (IRB).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
The participant has provided informed consent for the submission of the article to the journal.