
Abstract
Background
Opioids have been an integral part of pain management for many years. With the increasing global burden of opioid dependence, the development of non-opioid therapies provides a new framework for pain management. Suzetrigine is a stepping stone toward non-opioid treatment methods.
Purpose
To summarize the pharmacological profile, preclinical and clinical trial evidence of suzetrigine in opioid-free pain management.
Materials and Methods
Published research, clinical trial data, and review articles from PubMed, Cochrane, and Google Scholar were reviewed from January to July 2025. The findings have been consolidated in this article to summarize them in relation to suzetrigine to provide an overview from pharmacological and clinical perspectives.
Results
In vitro and in vivo studies demonstrated the voltage-gated sodium channel, NaV1.8, selective property of the drug. Vertex Pharmaceuticals has conducted four randomized controlled trials (two Phase 2 and two Phase 3 [NAVIGATE-1 and NAVIGATE-2]) and one open-label trial to assess the efficacy and safety profile of suzetrigine. Significant pain reduction was observed with suzetrigine compared to that with placebo. Nausea was the most common adverse effect observed with the drug; however, the drug was otherwise well tolerated and showed low abuse potential. The United States Food and Drug Administration approved suzetrigine for the treatment of moderate to severe acute pain on January 30, 2025. Several trials for the application of the drug in the management of chronic pain are ongoing, among which some have reported promising results. This review summarizes the pharmacological profile of suzetrigine, its future perspectives, current trends, and potential changes in pain management.
Conclusion
Suzetrigine is a first-in-class non-opioid analgesic and NaV1.8 inhibitor, approved only for the treatment of moderate to severe acute pain. With a less addictive nature and good safety profile, its potential in the management of conditions with chronic pain is yet to be elucidated from clinical trials with a long-term follow-up period.
Introduction
Pain is a universal complaint across all age groups.1–3 Its mechanism is influenced by diverse cultural and historical perspectives.2, 3 Being subjective, pain is difficult to quantify. The International Association for the Study of Pain (IASP), established in 1973, advanced the understanding of pain mechanisms. 2 The IASP, in 1979, defined pain as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage.”4, 5 With growing scientific knowledge, ICD-11 introduced a new pain classification (2013), later adopted by the WHO in 2019. 5 In 2020, viewing pain as a global healthcare issue, the IASP revised the definition as “an unpleasant sensory and emotional experience associated with, or resembling that associated with, actual or potential tissue damage.”5–7 Several theories explain the pain neuromatrix, including nociceptive component and its translation to Gate Control Theory, Bayesian Model, High-order, Global-workspace, and Information Theory. 4 Pain occurs when internal or external stimuli are transduced into nerve impulses via nociceptors and mechanoreceptors supported by ion channels.4, 8 Although the initial focus was on the central nervous system (CNS), the role of the peripheral nervous system in initiating and modulating pain is now being recognized. 4 Opioids remain a mainstay, but discovering voltage-gated sodium channel expression in the peripheral nervous system9–12 led to the development of newer analgesics, among which NaV1.8 expression and inhibition are clinically significant.
WHO Guidelines
Classifying pain as acute or chronic helps to identify the etiology. 3 Evidence of endogenous neurochemical reactions, opiate receptors, and psychological factors supports customized pain management.3, 7 Following international expert discussions, WHO introduced the three-step analgesic ladder, a guideline for cancer-related pain relief in 1986 for adults13, 14 and in 1998 for children. 13 The first step addresses mild pain, and moderate and severe persistent pain subsequently. 14 In 1996, a fourth step was added for acute pain. 14 Downward and upward approaches were formulated for acute and chronic pain, respectively, with analgesic platform or trolley methods incorporating non-pharmacological strategies.15–18 A “reverse pain ladder” for discharge planning after treatment for acute pain has also been proposed. 19
Trends in Pain Management and New Analgesics
A multimodal strategy is essential for all age groups and pain types.13,17,20–22 Targeting different levels of the pain pathway enables a non-opioid approach, especially in the peripheral nervous system.8, 23 Voltage-gated sodium channel blockers emerged from understanding nociception and ion channels in pain signaling.9–11,24 Neurotoxins, such as tetrodotoxin (TTX), act on tetrodotoxin-sensitive (TTX-S) subtypes NaV1.1, NaV1.2, NaV1.3, NaV1.4, NaV1.6, and NaV1.7.10, 25 TTX lacks affinity for myocardial NaV1.5 at therapeutic doses.
25
Intramuscular and intravenous administration showed a narrow therapeutic index, while oral administration of local anesthetics and vasoconstrictors widened the therapeutic index.
25
Analgesic effects have been demonstrated on neuropathic, inflammatory, acute, chronic, visceral, and malignant pain models at sublethal doses.
25
Other neurotoxins, such as neosaxitoxin, produced minimal nerve conduction block.10, 26 Trials confirm potentiation of the local anesthetic effect of neosaxitoxin when coadministered subcutaneously with bupivacaine and epinephrine.
26
Batrachotoxin, peptide-based venom, or toxins from scorpions, spider-like protoxin, huwentoxin, and pyrethroid also modulate NaV function.
26
Lidocaine and fentanyl were repurposed for their analgesic effect. First-generation NaV modulators were non-selective and caused state-dependent block, binding to the inner pore of the channel. This includes lidocaine, phenytoin, and anti-arrhythmic drugs. Due to systemic side effects from lack of selectivity, first-generation drugs are not preferred for chronic pain management.
10
The second-generation drugs include ralfinamide and lacosamide. Ralfinamide blocks N-type Ca2+ channels and N-methyl-
Current analgesics for chronic pain, such as opioids, dorsal root ganglionic stimulation (DRGS), peripheral nerve stimulation (PNS), and telemedicine, act on the CNS. 27 Drugs targeting NaV1.1, NaV1.3, and NaV1.7 aim to overcome CNS-associated adverse effects like sedation, dependence, euphoria, and addiction.8, 24, 25
Selective NaV1.7 inhibitors, such as PF-05089771 and CNV1014802, and NaV1.7, NaV1.8, and NaV1.9 blockers, such as DSP-2230 (ANP-230), are currently under clinical trial. 10 NaV1.8-specific drugs such as A-803467, VX-128, and VX-150 failed with serious adverse reactions in Phase 1 studies. 28 VX-548 performed better, and after two successful Phase 3 clinical trials, the Food and Drug Administration (FDA)-approved VX-548 as suzetrigine for acute pain management on January 30, 2025. Other NaV1.8 inhibitors are in Phase 1 and Phase 2 clinical trials. 29 Suzetrigine is challenged by cost and limited efficacy compared to gene therapy targeting NaV1.7 and NaV1.8. 30
Opioid Epidemic
Therapeutic and illicit opioid misuse remains a major concern.7, 26 Long-term use resulted in relapse and remission of opioid use disorder (OUD).26, 31 From 1989 to 2009, opioid use peaked globally. 19 An observational epidemiological study conducted based on the 2019 Global Burden of Disease Study showed an escalation in the incidence, prevalence, and mortality of OUD among adolescents and young adults from 1990 to 2019. 32 In 2019, globally, 43,447 adolescents and young adults died from OUD. 32 Men predominated in high sociodemographic index countries. 32 However, after 2014, the incidence in females increased. The World Drug Report from the Centers for Disease Control and Prevention (CDC) reported a 38% increase in mortality from opioid overdose, with 70% of drug-overdose deaths due to opioids. 33 Although opioid dependence can be treated with naloxone, naltrexone, methadone, and buprenorphine, measures to prevent opioid addiction are important.19, 26 Psychosocial factors influence relapse or opioid dependence recurrence. 34 Both genetic and environmental factors predispose opioid addiction. 35 Neurocircuit dysregulation involving reward-stress function contributes to OUD. 35 Genomic biomarkers are being explored to enable personalized risk prediction in chronic opioid users.33, 35 A recent study integrated genome-wide association study (GWAS) traits with in silico analytic methods, such as protein–protein interaction (PPI) and enrichment analysis (EA), identified 50 genes linked to pain, opioid use, and inflammation, highlighting µ-opioid receptor (OPRM1), dopamine receptor (DRD2), glutamate receptor (GRIN2B), GPR98, and APOE with implications for neurological disorders and lipid metabolism. 36 It proposed a combination of high-density lipoprotein (HDL)-boosting drugs and dopamine-boosting regulators, such as KB220, to reduce relapse. 36 Therefore, opioid addiction can be predicted prior to usage. Another study used next-generation sequencing (NGS) to identify genetic variants (single-nucleotide variants) predisposing OUD, using RNA-sequencing data from postmortem ventral midbrain tissue of chronic opioid users. Out of eight genes, NRXN3 (Neurexin 3), involved in synaptic transmission, had the highest number of variants in chronic opioid users. 33 Thus, non-opioid analgesics are promising alternatives for acute pain.
Methodology
Published research and review articles related to pain were searched from PubMed, Cochrane, and Google Scholar. In addition, clinical trial data from NCT04977336, NCT05034952, NCT05660538, NCT06176196, NCT05553366, NCT05558410, and NCT05661734 were reviewed. Keywords used for the search were pain, suzetrigine, NaV, opioids, and analgesia. Only articles published from January to July 2025 were included in this review article. Both preclinical and clinical studies were included. The results were consolidated for a pharmacological and clinical overview of suzetrigine.
Results
This section summarizes preclinical and clinical trial data of suzetrigine. This includes pharmacokinetics, efficacy, and safety outcomes from the studies.
Preclinical Study (In Vitro and In Vivo)
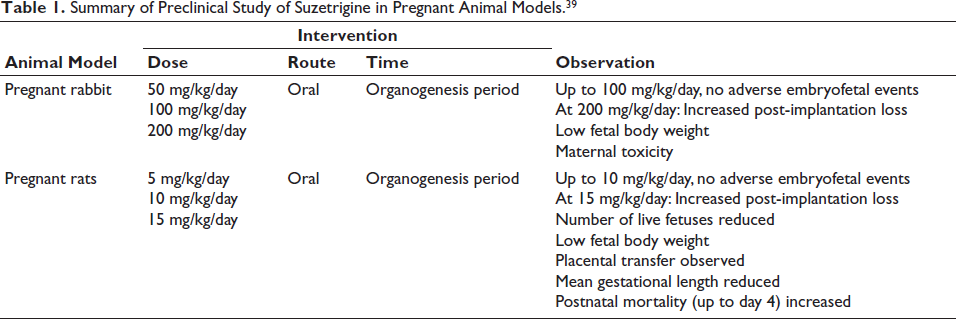
Preclinical in vitro and in vivo studies demonstrated high selectivity of suzetrigine. 37 Its specific binding site is on the voltage-sensing domain 2 (VSD2) in the closed state of the channel, confirmed by introducing NaV1.8’s VSD2 into NaV1.2 using a domain-swapping strategy. 37 NaV1.2, unresponsive or insensitive to suzetrigine, showed sensitivity to the drug when attached to VSD2 and no other voltage-sensing domains (VSD1, VSD3, and VSD4) of NaV1.8. Thus, VSD2 plays a key role in the drug sensitivity of NaV1.8.6, 20, 37 Direct binding to VSD2 was confirmed by using radiolabeled suzetrigine. Some studies suggested that the binding site is based on the extracellular loop of S3–S4 segments of VSD2, while others point out the KKGS sequence—a unique amino acid motif in the S3–S4 extracellular loop in NaV1.8—to confer sensitivity in NaV1.8 to suzetrigine through experiments.37, 38 VSD2 regulates pore opening. Unlike anesthetics that bind to the inner pore of the channel or open state, suzetrigine interferes with the movement of VSD2 in its resting state or conformation to modify the gating mechanism externally. Eventually, NaV1.8 is stabilized in the closed or resting state, inhibiting further activation or opening to nerve signals. This causes allosteric and tonic state-dependent inhibition rather than a simple channel-blocking mechanism.31, 37 To find out NaV subtype selectivity and potency of suzetrigine, electrophysiological studies were performed using cell lines with stable or transient overexpression of NaV channels such as Chinese hamster ovary (CHO), Human embryonic kidney (HEK), and ND7/23 (rodent neuroblastoma fusion). 37 The number of action potentials observed before and after the administration of suzetrigine, using the manual patch voltage clamp method, was used to assess the inhibition of suzetrigine effects. 37 No cardiovascular and respiratory adverse effects were observed in a single-dose Latin square design study conducted in monkeys. In vivo studies in rats and monkeys and clinical trials for CNS effects, adverse effects, and abuse potential favored a lower chance of addiction, despite crossing the blood–brain barrier, due to lack of NaV1.8 expression in the CNS. 37 No behavioral changes or physical dependence were observed. However, the long-term safety profile and intensity of inhibition in correlation with efficacy have yet to be established through further clinical studies. Preclinical studies on its effects during pregnancy showed the findings presented in Table 1. In a rat study, preimplantation loss was attributed to the high sensitivity of rat progesterone receptors, unlike those in humans. 39 In addition, the effect on fertility was reversible. Stopping the drug 4 weeks prior showed no preimplantation loss. Preimplantation loss was observed only when the drug was administered before mating or until gestational day 7. To assess the systemic toxicity of suzetrigine and M6-SUZ, a repeat-dose study was done in rats and monkeys for 91 days and 28 days. 39 In vitro Ames, in vitro, and in vivo micronucleus assays were negative for mutagenicity and carcinogenicity of suzetrigine. 39
Summary of Preclinical Study of Suzetrigine in Pregnant Animal Models. 39
Pharmacokinetics of Suzetrigine
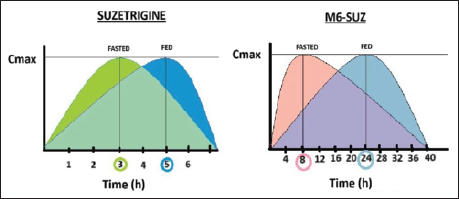
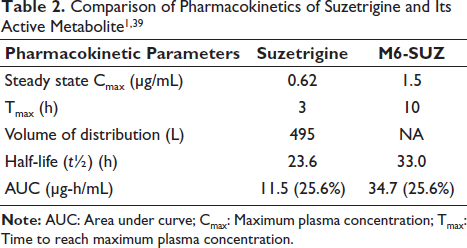
In vitro electrophysiological assays used manual patch voltage clamp experiments in isolated human and monkey dorsal root ganglia neurons to determine the IC50 as a measure of drug potency. 37 IC50 was 0.68 nM in humans, 0.75 nM in monkeys, and 56 nM in rodents, reflecting markedly lower rodent NaV1.8 sensitivity. This 80-fold difference explains why rodent in vivo models were not used. In vitro human tissue studies demonstrated that M6-SUZ, the active metabolite, also blocks NaV1.8. CYP3A enzymes form M6-SUZ. 40 Rat liver microsomes: M1 (O-desmethyl) and M2 (N-oxide), with M1 predominating in males, demonstrated sex-dependent metabolism in rats, unlike in humans.37, 40 This interspecies and sex-dependent metabolic variation influences the pharmacokinetics of the drug, limiting the translation of preclinical data to humans. The pharmacokinetic studies in rats, monkeys, and humans used liquid chromatography–mass spectrometry (LC–MS) bioanalytical assay to quantify the plasma concentrations. 1 The Cmax was 0.62 µg/mL, and Tmax was 3 h during the fasting state and 5 h during the fed state.37, 39 This explains the delayed onset of action in the fed state in Figure 1. Suzetrigine shows 99% plasma protein binding, a volume of distribution (Vd) of 495 L, and a half-life (t½) of ~23.6 h, reducing its clearance to ~3.9 L/h.20, 37, 39 Suzetrigine is eliminated by oxidative metabolism, with 49.9% excreted in feces (M6-SUZ and suzetrigine) and 44% in urine (only M6-SUZ). 39 Bioavailability remains unreported. 39 Table 2 compares the pharmacokinetic parameters of suzetrigine and M6-SUZ.
Clinical Trials
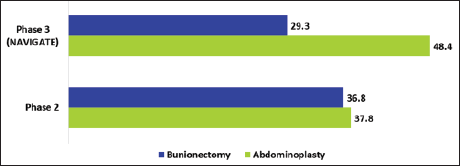
Five clinical trials—two Phase 2 and three Phase 3 (NAVIGATE-1, NAVIGATE-2, and single-arm study) trials—evaluated the efficacy and safety of suzetrigine.41–43 The results are summarized in Table 3. Except for the Phase 3 single-arm study, all trials were randomized, double-blind, active- and placebo-controlled studies.41, 42 Abdominoplasty and bunionectomy served as soft- and hard-tissue pain models. The endpoints of NAVIGATE-1 and NAVIGATE-2 were the same, reporting efficacy using the sum of the pain intensity difference over 48 h (SPID48) as the primary endpoint and safety as the secondary endpoint. 41 The SPID48 was expressed as the least square (LS) mean difference. The results are presented in Figure 2. Higher SPID48 values indicate better pain relief. The open-label study included surgical pain from orthopedic procedures, most commonly ligament repair, arthrodesis, followed by liposuction, mammoplasty, nasal septoplasty, turbinoplasty, inguinal hernia repair and non-surgical pain from arthralgia, limb pain, and strain. The safety profile was consistent with controlled trials. 39 Overall, suzetrigine demonstrated superior efficacy compared to placebo at high doses during the postoperative period. 44 In a phase 3 abdominoplasty trial, suzetrigine produced better pain relief compared to placebo—119 versus 480 min in abdominoplasty and 240 versus 480 min in bunionectomy. 39
Least Square (LS) Mean Difference in SPID48 (Suzetrigine vs. Placebo).1, 39 The More the Difference in LS Mean, the Better the Pain Reduction. Suzetrigine Showed Better Pain Relief in Abdominoplasty in Both Phase 2 and 3 Studies.
Summary of Clinical Trials Conducted With Suzetrigine.
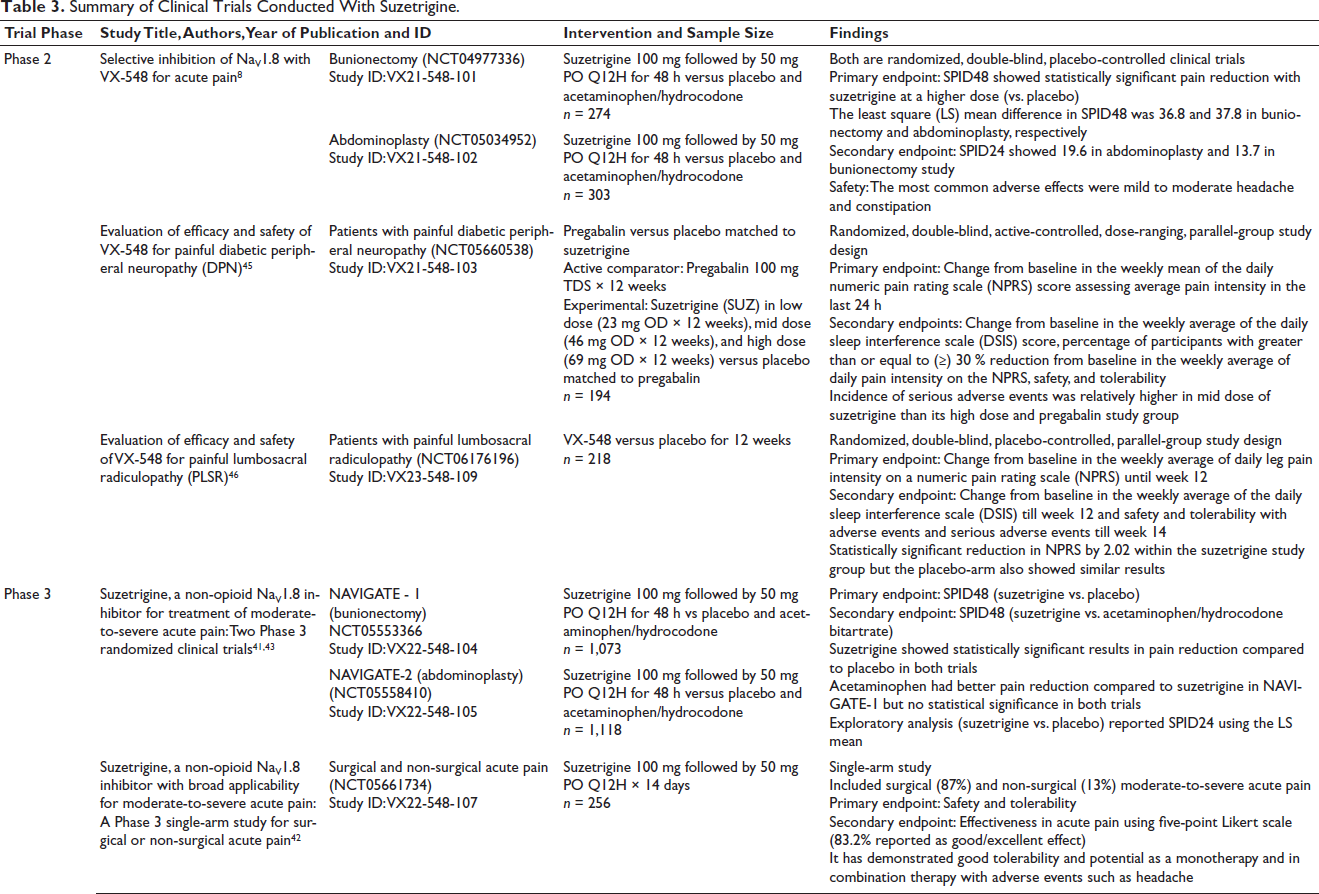
These studies demonstrated the superiority of suzetrigine over placebo, but only at high doses, limited to 48 h post-surgery and a maximum of 14 days. Limited evidence on the dosage of active comparator (acetaminophen-hydrocodone) questions the efficacy of suzetrigine in a real-world clinical setting for chronic pain. 44
Promising results from initial studies have promoted clinical trials for more commonly addressed chronic pain conditions, such as diabetic peripheral neuropathy and lumbosacral radiculopathy.45, 46 In Phase 2 study of lumbosacral radiculopathy, suzetrigine did not outnumber the placebo. In Phase 2 study of diabetic peripheral neuropathy, high-dose suzetrigine and placebo showed a better safety profile than mid-dose suzetrigine. A Phase 3 study on diabetic peripheral neuropathy is currently ongoing. As suzetrigine failed to outnumber placebo in a Phase 2 trial with lumbosacral radiculopathy, approval for advancement to Phase 3 is pending. As suzetrigine has been approved only for the management of acute pain, its analgesic potential in chronic pain depends on the awaited outcomes of ongoing trials and further studies.
Safety Profile
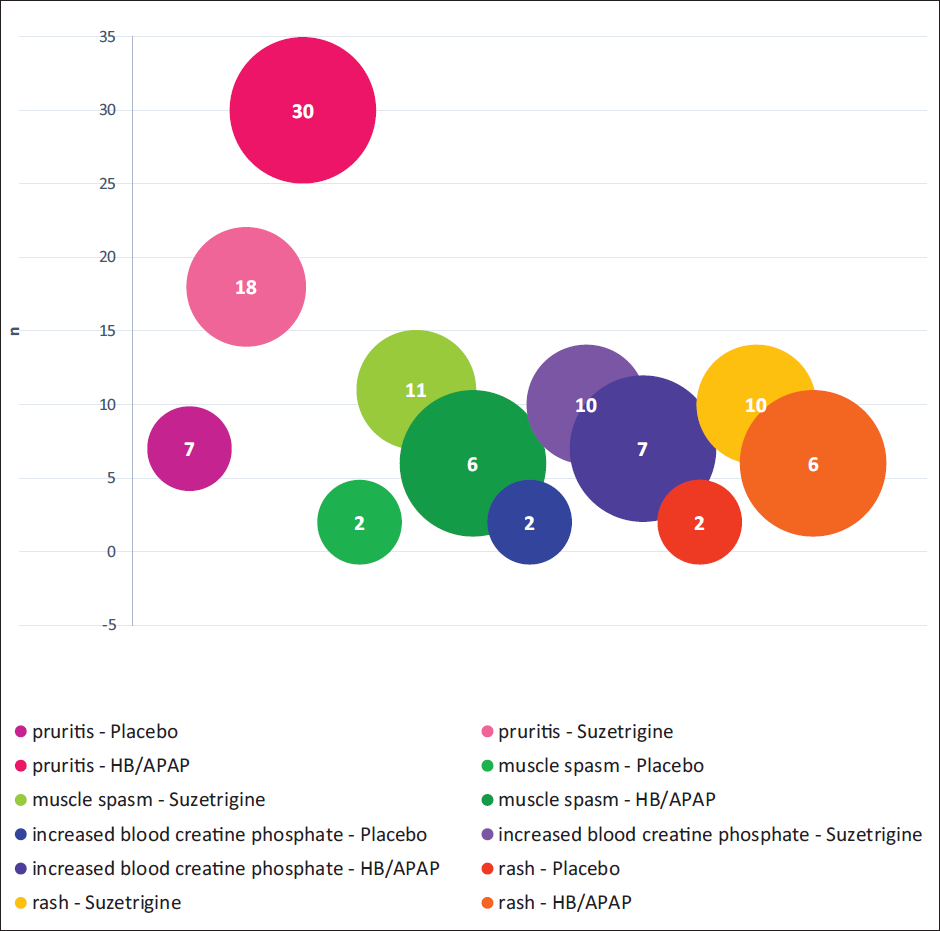
The trials demonstrated suzetrigine to be a safe drug with no abuse potential in the population studied for a particular dosage. 39 The most common adverse effect of suzetrigine in Phase 3 trials (NAVIGATE-1 and NAVIGATE-2) was nausea or vomiting (20% in abdominoplasty and 9% in bunionectomy patients). The other adverse effects included pruritus (18%), muscle spasm (11%), increased blood levels of creatine phosphokinase (CPK) (10%), and rash (10%). 39 Nearly 2.9% of participants in the suzetrigine group had CPK levels three times higher than the normal upper limit. Approximately 2.5% of participants treated with suzetrigine in the NAVIGATE-1 and NAVIGATE-2 trials had a reduction of ≥25% and <50% in estimated glomerular filtration rate (eGFR), respectively. A reduction in eGFR was also reported in the open-label Phase 3 trial; however, the values returned to baseline during follow-up. Although there was no adverse event due to this change, the reason was not stated; however, it is recommended to avoid the drug in patients with renal impairment or dysfunction. In addition, the incidence of adverse effects of suzetrigine was lower than that of hydrocodone bitartrate/acetaminophen and placebo. Adverse effects were more common in the acetaminophen treatment group than in the placebo group. Figure 3 shows the incidence of adverse effects in the three treatment arms. In addition, the safety profile of suzetrigine in the Phase 3 open-label trial was consistent with that in NAVIGATE-1 and NAVIGATE-2.
Adverse Reactions Reported in Two 48-h Trials in Moderate to Severe Acute Pain (Trials 1 and 2, Pooled).1, 39 The Most Common Adverse Effect was Pruritus Followed by Muscle Spasm, Increased Blood Creatine Phosphate, and Rash in All Three Treatment Groups. Hydrocodone/Acetaminophen (HB/APAP) Group Had the Highest Incidence of Pruritus, But Suzetrigine Group Showed Increased Incidence of Other Side Effects.
Preclinical toxicity studies have demonstrated the potential of suzetrigine to affect fertility, pregnancy, and fetal growth in pregnant animal models. 39 Despite the physiological differences between animal models and humans, the use of suzetrigine should be avoided. In NAVIGATE-1 and NAVIGATE-2, the change in mean systolic and diastolic blood pressure in the suzetrigine group compared with the placebo group was not clinically significant.1, 39 Most of the adverse events during the trial were mild and self-limiting. The incidence of serious adverse events and adverse events leading to discontinuation was minimal in this study. No specific antidote has been developed for drug overdose or drug toxicity. Supportive treatment with monitoring of clinical status and vitals is recommended. 39
Discussion
The trial outcomes demonstrate the efficacy of suzetrigine in the management of moderate to severe acute pain of surgical and non-surgical etiology only. With good safety outcomes, its potential in chronic pain management is being studied in the ongoing trials. The FDA-approved therapeutic uses, dosage, route of administration, drug and food interactions, and application in special populations are discussed below.
Role of NaV1.8 in Pain Signaling
Voltage-gated sodium channels regulate the cell membrane potential and vary by alpha subunit expression, coded by the SCN gene. They are expressed in the CNS, peripheral nervous system, or both. NaV1.8 channels are mainly expressed in the dorsal root ganglion, nociceptors, and pain pathway signaling, but not in the CNS.37, 47, 48 Initial studies found NaV1.8 to be tetrodotoxin-resistant (TTX-R), with selective expression in dorsal root ganglion neurons of small-diameter like C-type nociceptive fibers.28, 49 Electrophysiological studies show a positive correlation between NaV1.8 expression and both the duration and amplitude of action potential, suggesting that NaV1.8 is involved in the nociceptor and pain signaling pathway. 50 Immunohistochemical analysis revealed the highest immunoreactivity in Aδ fibers, followed by C-type and Aα/β nociceptive fibers. Thus, NaV1.8 is involved in chronic pain.47, 50 Nociceptors express NaV1.8 more than mechanoreceptors.23, 50 NaV1.8 functions by cyclical opening and closing, called the gating mechanism. Among all NaV subtypes, NaV1.8 requires strong depolarization or a high voltage threshold for activation or opening. For longer, stable Na+ influx, a prolonged open state gives a sustained action potential or pain signal transmission, that is, NaV1.8 inactivation is slow. 37 Subsequent voltage-gated K+ efflux restores the resting membrane potential. 11 It is active regardless of NaV subtypes. 51 This explains its role in the development of neuropathic pain. 52 Rapid recovery from inactivation causes repetitive pain signals. 9 Gain or loss of function mutation in NaV1.8 predisposes chronic pain.38, 48 Thus, NaV1.8 plays a role in both nociception and chronic pain.9, 48
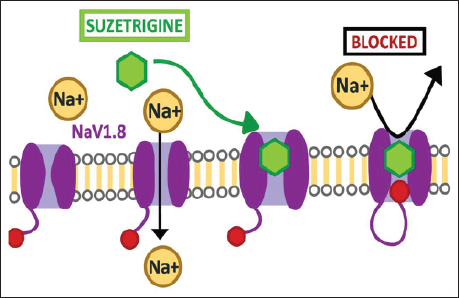
Mechanism of Action
Suzetrigine selectively binds to VSD2, producing NaV1.8 blockade and preventing sodium influx needed for nociceptive signaling between neurons by stabilizing the channel in a closed or inactive state.37, 39 This mechanism is illustrated in Figure 4.
Dosage and Administration
Suzetrigine is available as a 50 mg oral tablet. The recommended loading dose is 100 mg, to be taken in a fasting state, either 1 h before or 2 h after a meal. The maintenance dose of 50 mg is started 12 h after the loading dose, and subsequent doses are repeated every 12 h. The treatment duration is a maximum of 14 days, as studies have not covered beyond this duration of intervention. In patients with moderate hepatic impairment, especially Child–Pugh Class B, the fifth and subsequent doses are given at 24-h intervals. In case of missing a single dose or ≥2 doses, patients were advised to take the missed dose (one tablet for a single missed dose and two tablets for skipping ≥2 doses) and continue the next scheduled dose at the recommended time. For patients with hepatic impairment or under CYP3A inhibitor, on missing a dose, if the next scheduled dose is within or equal to 6 h, the missed dose is to be skipped and the next scheduled dose is to be taken. If the time to take the next dose is more than 6 h, it is advised to take the missed dose and continue with the next scheduled dose.1, 39 In patients with eGFR 15 to <60 mL/min, pharmacokinetic parameters were unaltered. Given that patients with renal impairment or an eGFR less than 15 mL/min were not included in the study, it is advised to avoid using suzetrigine in this population as a precautionary measure. 39
Clinical Indications
Suzetrigine has been used for the management of moderate to severe acute pain of both surgical and non-surgical etiologies, backed by evidence from clinical trials. 39 As the study period in the trials was up to a maximum of 14 days, the chronic use of the drug and its potential for harm or more benefits are yet to be concluded. Trials have been extended to common chronic pain conditions, such as lumbosacral radiculopathy and diabetic peripheral neuropathy, and the results of these trials will further test the long-term use of suzetrigine. Based on their observations, some studies suggest the use of suzetrigine with other non-steroidal anti-inflammatory drugs (NSAIDs) as a combination therapy for better pain relief. 51
Drug and Food Interactions
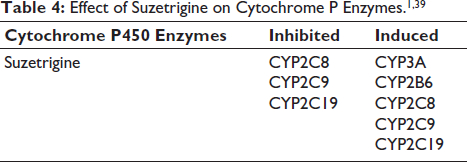
As a CYP3A4 inducer, suzetrigine induces the metabolism of CYP3A4 substrates, such as direct oral anti-coagulants (DOACs). This raises the risk of thromboembolism when both drugs are administered concomitantly, necessitating dose monitoring or avoidance of coadministration. 53 The effects of suzetrigine on cytochrome P enzymes are listed in Table 4.
On administering 100 mg of suzetrigine with high-, moderate-, and low-fat meals, the plasma concentration of the drug and its metabolite M6-SUZ declined, unlike in the fasting state. However, the fed state is predicted not to affect subsequent maintenance doses due to no change in the systemic response. The effect of food on the time to onset of action is shown in Figure 1. Based on the delayed Tmax and initial plasma concentration with moderate- and high-fat meals, the consumption of the loading dose of 100 mg in the fasting state, that is, 1 h before or 2 h after food. Subsequent doses can be taken with or without food. Consumption of grapefruit-containing foods should be avoided as they inhibit CYP3A4, which metabolizes suzetrigine.1, 20, 39 Overall, precautions should be taken, such as avoiding the concomitant use of strong or moderate CYP3A inhibitors, CYP3A substrates, and hormonal contraceptives containing progestin. Additional non-hormonal or alternative contraceptive methods will be needed during treatment and for 28 days after stopping suzetrigine. Precautions related to the dose in patients with hepatic impairment or dysfunction are mandated. 39
Role in Special Populations
A preclinical study showed placental transfer of suzetrigine in pregnant animal models, but no embryo-fetal effects were observed up to a dose of 100 mg/kg. Furthermore, there was no impact on learning, memory, or sexual maturation in newborns up to 15 mg/kg. As the drug was detected in animal milk, it is also likely to be present in human breast milk. The drug-associated risk of miscarriage, maternal complications, or congenital anomalies is unknown from the trials conducted so far. 39
The trial participants were primarily middle-aged. The trial included only adults aged >18 years. The pediatric population was not included in any of the studies; without further evidence, the drug is not recommended for this group. The pharmacokinetic parameters among participants aged 18–75 years showed that age had no clinical relevance to suzetrigine. 39
In patients with hepatic dysfunction or impairment, a low dose is recommended for moderate severity, that is, Child–Pugh Class B. Suzetrigine should be avoided in severe hepatic impairment, that is, Child–Pugh Class C. Suzetrigine should be avoided in patients with eGFR <15 mL/min. 39
Conclusion
Suzetrigine is a first-in-class NaV1.8 inhibitor approved for the management of moderate-to-severe acute pain. Given its low abuse potential, it is a promising alternative in addressing the global opioid epidemic. Based on clinical trials, this drug is used for the management of moderate to severe acute pain of both surgical and non-surgical etiology. The recommended loading dose is 100 mg, 1 h before or 2 h after food, followed by a maintenance dose of 50 mg, every 12 h. The maximum treatment duration is 14 days. The safety profiles of all clinical trials yielded similar results. Although trials to date have evaluated the efficacy and safety with respect to acute pain, further studies are required to explore its long-term safety and potential as an analgesic for chronic pain. This review suggests the need for further studies on suzetrigine in the pediatric, geriatric, and renal impairment populations.
Footnotes
Acknowledgments
No additional individuals other than the authors contributed to the preparation of this article.
Abbreviations
Declaration of Conflict of Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Not applicable.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors gratefully acknowledge the financial support by SRM Medical College Hospital and Research Centre, Faculty of Medicine and Health Sciences, SRM Institute of Science and Technology (SRMIST), Kattankulathur, Chengalpattu, Tamil Nadu, India, for bearing the defrayed costs of publishing this article.
Informed Consent
Not applicable.