
Abstract
Background
The brain, through the sensory action of neurons situated in the hypothalamus and brainstem, plays a pivotal role in the homeostatic regulation of glucose metabolism. The brain senses changes in nutrients and hormones. It triggers negative feedback responses such as stimulating pancreatic insulin secretion, hepatic glucose production, and glucose/fatty acid metabolism in adipose tissue and skeletal muscle, and cellular glucose uptake, to restore glucose homeostasis. When glucose homeostasis is not tightly regulated, hyperglycemia may manifest, leading to diabetes mellitus (DM) and, in some cases, trickling into Alzheimer’s disease (AD).
Objectives
This review article aims to establish the link between DM and AD by describing the processes from the onset of glucose dysregulation to the resulting changes in the brain that trigger AD symptoms. Furthermore, this review provides an understanding of interlinked processes, and proposes the repurposing of DM drugs as candidates for AD.
Methodology
Literature search was conducted using search engines such as PubMed and Google scholar, with papers written in English were selected.
Results
Repurposing DM treatment towards AD is a promising area of research due to their pathogenically common features, which include metabolic dysfunction, insulin resistance, and inflammatory processes. This review highlights various diabetes treatments that could be possible molecular targets for AD treatment. Drugs such as glucagon-like peptide-1 (GLP-1) agonists, sodium-glucose co-transporter-2 (SGLT-2) inhibitors, and intranasal insulin delivery are being explored and have shown potential for improving outcomes in AD. Through this exercise, we envisage that drug candidates and existing drug targets could be realized, most especially for AD, where optimal treatment is still a challenge.
Conclusion
This article further summarizes a few DM treatment strategies that could be further investigated as drug targets to improve disease outcomes for AD patients.
Introduction
Several targets of the insulin machinery that may potentially catalyze the development of neurodegenerative diseases have been intensively studied and identified. Statistically, the risk of developing Alzheimer’s disease (AD) was 65% higher in persons with diabetes than in non-diabetic controls, as reported in the study. 1 This relationship is significant, such that AD has sometimes been referred to as the “diabetes of the brain” or “type 3 diabetes mellitus” (T3DM). 2 When glucose homeostasis is not maintained in the body, this can result in dysregulation of some metabolic, molecular, and cellular processes, which can lead to diabetic complications. In addition, these metabolic disturbances may lead to progressive structural and functional abnormalities in the brain, impairing neuronal signaling. 3 The brain senses peripheral metabolic signals through hormones such as insulin/leptin as well as nutrients to regulate glucose homeostasis. 4 As a response to these signals, the brain modulates and coordinates various aspects of metabolism, such as food intake, energy expenditure, insulin secretion, hepatic glucose production, and glucose/fatty-acid metabolism in adipose tissue and skeletal muscle to maintain glycemic control.4, 5 Accumulating evidence suggests that impaired cerebral glucose metabolism is an invariant pathological feature of AD. Chronic hyperglycemia results in accelerated formation and accumulation of advanced glycation end products (AGEs) in various tissues.6, 7 AGEs, although reported to increase during the normal aging process, play a critical role in the pathogenesis of diabetic complications and neurodegenerative disorders, including AD. 8 The activation of receptors for advanced glycation end products (RAGE) by AGEs in a variety of settings triggers rapid generation of reactive oxygen species (ROS) and the upregulation of inflammatory pathways. This can ultimately result in diabetic complications, amplification of inflammation, and a myriad of consequences. 9 In addition, the excess accumulation of amyloid beta (Aβ) plaques in the brain establishes a vicious cycle of impaired brain insulin signaling, inflammation, and oxidative stress processes that promote neurodegeneration in the AD brain. 10 Aβ accumulates in peripheral tissues such as the pancreas and can induce insulin resistance in the liver, which suggests that not only can insulin resistance promote Aβ accumulation, but also the reverse could occur. 11 Several studies have reported observations supporting the bidirectional relationship existing between AD and diabetes mellitus (DM). One study found a significantly lower cognitive performance among diabetic patients compared to healthy controls after a 4-year follow-up period. 12 Another cross-sectional study discovered that subjects with T2D performed worse in all cognitive domains than those with normal glucose metabolism. 13 Consistent with these findings, a recent meta-analysis of 144 prospective studies identified a 1.25–1.9-fold increase in cognitive impairment and dementia in patients with diabetes. 14 Therefore, by understanding and acknowledging the existence of these overlapping pathogenic features between AD and DM, we can target these two diseases holistically and find a much-anticipated effective treatment strategy with the aim of improving therapeutic outcomes for the patient and giving them the best quality of life. In this review, we intend to explore the linkage between diabetes and AD. Most importantly, it explores how the understanding of such a relationship can be harnessed for the management of AD at an etiological root.
AD Pathophysiology
AD is a degenerative disorder that involves the progressive and irreversible loss of neurons in regions of the brain. The etiology of AD has been linked to several mechanisms. The amyloid cascade hypothesis is believed to play a significant role in the neuropathology of AD. 15 This hypothesis suggests that the dysregulation of amyloid precursor protein processing that occurs in AD development at an early stage is associated with the overproduction of Aβ42, which clumps together to form plaques between neurons and disrupt cell function. 16 As the plaque “matures,” the Aβ peptides reorganize into β-pleated sheets and fibrillize into neuritic plaques. 17 The formation of these neuritic plaques results in oxidative injury, which drives neuronal degeneration and cognitive decline in AD patients. 18 Another brain structure, tau, is a microtubule-associated protein of the cytoskeleton best known for its role in stabilizing neuronal microtubules. Structurally, tau is a natively unfolded protein, highly soluble, and shows little tendency for aggregation. 19 In pro-oxidative states and with impaired glucose metabolism, hyperphosphorylation of tau and its dimerization in vivo are facilitated, potentially leading to cross-linking and the formation of helical filaments and, subsequently, neurofibrillary tangles, cytoskeletal abnormalities, axonal transport disorders, synaptic loss, and ultimately dementia. 20 The cholinergic hypothesis, which describes another pathway to AD pathophysiology, describes the role of acetylcholine (ACh), a neurotransmitter involved in the conduction of nerve signals in the central nervous system (CNS) and peripheral nervous system. 21 ACh mediates communication at the neuromuscular junctions and plays a role in memory, learning, and neuroplasticity, which is the brain’s ability to alter the structure of its neural network, allowing it to form neural connections. In the peripheral nervous system, ACh regulates a variety of tissue and organ functions, including the maintenance of glucose and cellular homeostasis. 22 Acetylcholinesterase (AChE) is the primary enzyme responsible for the hydrolytic metabolism of the ACh into choline and acetate, which results in the termination of signal transmission between nerve cells. 23 Ultimately, synaptic activities become altered, as well as the organ functions dependent on the activity of ACh, which result in AD and systemic diseases such as diabetes and cardiovascular disease.
Current AD Treatment Strategies
Two main drug classes have been approved for the treatment of AD. Cholinesterase inhibitors (CIs) are approved for dementia of the Alzheimer’s type in the mild-to-moderate stage. The second class is the N-methyl-
CIs
CIs, which include donepezil, rivastigmine, and galantamine, act by increasing the concentration of ACh at the synapse by binding to and inactivating these cholinesterases. Several studies have demonstrated that the cholinergic system plays a role in the processing of memory and learning, and the modulation of acquisition, consolidation, and retrieval of memory, to mention a few. 24 CIs also increase ACh activity in the peripheral nervous system, which contributes to the side effects commonly experienced. Anorexia, convulsions, and insomnia are some of the side effects experienced with the use of these classes of drugs.
NMDA Antagonists
N-Methyl-
While this class of anti-Alzheimer drugs represents the best currently available pharmacological treatments for AD, they have relatively insignificant overall effects and do not alter the course of the underlying neurodegenerative process. Hence, there is a need to identify newer drug targets for AD treatment.
Beta-amyloid Modulators
The conventional approaches targeting Aβ focus on three main therapeutic strategies. First, the reduction of Aβ peptide production via inhibition of β- and γ-secretases. Another approach is the use of amyloid anti-aggregates, which inhibit Aβ-peptide aggregation, for example, a synthetic glucosaminoglycan, 3-amino-1-propanesulfonic acid, and Colostrinin. Finally, compounds that facilitate the clearance of amyloid aggregates and deposits, for example, bapineuzumab and solanezumab, are the two monoclonal antibodies that have reached the most advanced stages of clinical development.26, 27 Aducanumab, another monoclonal antibody, is currently approved for its ability to reduce the number of Aβ plaques in the brain, although its approval seems controversial due to mixed efficacy results. 28 Lecanemab is the most recent Food and Drug Administration (FDA)-approved molecule, with promising potential. 29
Diabetes to Alzheimer’s Link: The Dysregulation in Glucose Metabolism
The human brain utilizes glucose as its main source of energy; therefore, the regulation of glucose metabolism is essential for brain physiology, including the generation of action potentials, communication between neurons, oxidative stress management, and the synthesis of neurotransmitters. Therefore, any disruption of normal glucose metabolism forms the pathophysiological basis for many brain disorders. Glucose is transported across the cell membranes by facilitated diffusion across the blood–brain barrier (BBB), mediated mainly by glucose transporter proteins GLUT-1 and GLUT-3. 30 GLUT-1 is responsible for glucose uptake across the BBB endothelial cells and into astrocytes, while GLUT-3 facilitates the uptake of glucose into neurons. 31 Most glucose uptake and metabolic processes by the brain are independent and therefore are believed not to be regulated by insulin signaling. However, insulin plays a critical role in cognitive behavior and the regulation of metabolic signaling pathways such as hepatic glucose production, lipolysis, lipogenesis, and the regulation of glucose homeostasis.32, 33 This highlights the interdependence between metabolic and cognitive functions in the human body; hence, any dysfunction in glucose metabolism, which is a key feature in DM, could result in cognitive dysfunction.
The existing links between diabetes-related pathologies and their debilitating effects on brain function have been established and studied. The next section describes the possible link between these diseases. At the molecular level, from the onset of glucose dysregulation, it trickles down a series of dysfunctional mechanisms that result in a disrupted nerve communication, which can ultimately lead to AD. The AD and DM linkage is a complex, multifactorial relationship; however, these complexities will be described in five main steps.
Insulin Deficiency on Hippocampal Function
Experimental reports have provided evidence on insulin receptors in the hippocampal tissues. The regulation of these receptors was shown to increase during spatial learning, therefore suggesting insulin involvement in cognitive functions. Further studies demonstrated that insulin regulates neural plasticity in the hippocampal tissue. 21 From a molecular perspective, insulin signaling, through PI3K/AKT, has a regulatory function on the NMDA and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors. NMDARs play a crucial role in synaptic transmission and synaptic plasticity, responsible for learning and memory.34, 35 Synaptic NMDARs are neuroprotective, and their dysfunction is directly involved, where they are overly stimulated and release glutamate in the synapses. Insulin has been reported to regulate these ionotropic gated channels, which facilitate calcium entrance through glutamate binding. The regulation of these receptor channels by insulin can be achieved at both transcriptional and phosphorylation for NMDA and AMPA receptors, respectively. 25 Chronic hyperglycemia associated with diabetes can cause overstimulation of the receptors, leading to excessive glutamate activity. Changes in either insulin signaling or insulin sensitivity in the hippocampus may alter molecular pathways involved in synaptic plasticity and adult neurogenesis. This will result in oxidative stress and inflammatory processes, which are toxic to neuronal activity.
Oxidative and Inflammatory Processes as a Response
The brain is an organ that requires a high oxygen demand to perform its activities properly. However, the brain has limited anti-oxidant substances and is rich in polyunsaturated fatty acids that are susceptible to peroxidation. 36 In addition, Aβ accumulation in the brain attracts immune system cells such as astrocytes, activated microglia, and macrophages, among others, which trigger an inflammatory response and promote an oxidative stress state. 37 The polyol pathway, often observed in DM, is considered the genesis of oxidative products that stimulate inflammatory processes.
Polyol Pathway
Under normoglycemic conditions, glucose oxidation produces energy in the form of adenosine triphosphate (ATP), nicotinamide adenine dinucleotide phosphate (NADPH), and ribose via the pentose phosphate pathway. However, in a hyperglycemic state, the polyol pathway is activated. 38 The activation of the polyol pathway results in the depletion of NADPH and the subsequent inability of glutathione reductase to use it for anti-oxidant purposes. Thus, the anti-oxidant capacity of the cell is compromised, leaving it vulnerable to oxidative stress and non-enzymatic glycation. The sorbitol produced within the neuronal cells or supporting cells can be detrimental as it can induce osmotic stress within the cells. Furthermore, the sorbitol can be converted to fructose, which is subsequently broken down to produce glyceraldehyde-3-phosphate (GA3P) and dihydroxyacetone phosphate (DHAP). 39 These metabolites have been reported to be precursors of advanced glycation end products and further activate the protein kinase pathway, which is central to inflammation. The polyol pathway and aldose reductase inhibitors have been sought for years, which has led to the discovery of alrestatin, benurestat, epalrestat, fidarestat, imirestat, lidorestat, minalrestat, ponalrestat, ranirestat, risarestat, sorbinil, tolrestat, zenarestat, and zopolrestat. Nevertheless, the clinical efficacy of this agent is controversial; hence, the FDA approval is still pending. By virtue of their neuroprotective effect, these drugs, through careful development and study, can perhaps be contested as therapeutic or adjuvant therapy for AD.
AGEs
The accumulated glucose and other monosaccharides in the blood and body tissues due to insulin resistance undergo non-enzymatic reactions with free amino acids, lipids, and protein molecules floating around in the cell. 40 Furthermore, the glycolysis operation is limited in diabetes due to mitochondrial disturbance. Glucose undergoes autoxidation, which produces glyoxal and methylglyoxal, which are the main precursors. Furthermore, GAP and DHAP can also undergo autoxidation and dephosphorylation, which also produce these substrates. Glyoxal and methylglyoxal can react with side chains of amino acids in proteins, thus forming what is known as AGEs.41, 42 These resulting adducts are also known as AGEs and can break down cells and tissues, which can trigger the immune response and inflammatory processes. AGEs activate RAGE. Increased RAGE expression and modified proteins can lead to mitochondrial damage and induce ROS, which promote oxidative stress, the formation of amyloid plaques, neurofibrillary tangles, and inflammation, which is a common feature in the AD brain. 43 AGEs and oxidative stress mechanisms are some of the major contributors to diabetic complications such as retinopathy, neuropathy, and nephropathy. 44 Aminoguanidine also known as pimagedine, is an investigational or prototype drug for AGEs formation inhibition. Despite its promise to avert diabetes-associated complications; however, its safety issues worked against it. However, its potential to alleviate AGEs production can still warrant the premise as a prototype for anti-AGEs tailored for AD.
Increased Expression of Matrix Metalloproteinases (MMPs) Proteins
The extracellular matrix (ECM) in the brain has emerged as an important reservoir of signaling molecules, which can influence synaptic plasticity, synaptogenesis, neurite outgrowth, and other processes occurring in the CNS. 45 MMPs are among the major modulators of the ECM and are involved in the proteolysis of ECM components, growth factors, cerebrovascular basement membrane, tight junction proteins, and cell surface protein receptors. 46 Systemically, hyperglycemia, via oxidative stress and AGEs, increases the expression of MMPs, resulting in alterations in vascular components, which have a direct implication for diabetic complications. 47 The development of diabetic peripheral arterial disease is mediated by the activation of enzymes that act on signaling molecules such as pro-inflammatory cytokines, cell surface receptors, cell–cell adhesion molecules, and the induction of oxidative stress. 48 In this context, Ilomastat, a synthetic inhibitor of MMPs, has shown promise in preclinical research related to AD due to its potential neuroprotective effects. 49 AD involves neurodegenerative processes like amyloid plaque accumulation, inflammation, and degradation of the BBB, which are associated with MMP activity. MMPs, particularly MMP-2 and MMP-9, are thought to contribute to neuronal damage, ECM degradation, and inflammation, potentially exacerbating Alzheimer’s progression.50, 51 Ilomastat’s ability to inhibit MMPs suggests it might help stabilize the BBB and mitigate neuroinflammation, reducing neuronal and synaptic damage.52–54 Research in animal models has indicated that inhibiting MMP activity can reduce amyloid-beta plaque formation and attenuate inflammation, offering a pathway to slowing Alzheimer’s progression. 55 However, despite its potential, Ilomastat has not yet advanced to clinical trials for AD, and more studies are needed to clarify its effects, safety, and potential as a therapeutic agent in humans.56–58 Succinctly, Ilomastat may represent a novel approach to targeting AD by modulating MMP activity and protecting neural structures. However, translating these findings into clinical use requires substantial further research.
Nerve Cell Damage from Inflammation
The pathophysiology of diabetic neuropathy is complex. Insulin signaling in the CNS promotes neuronal plasticity, synaptic density, and function, ensuring key processes underlying learning and memory are maintained. In a diabetic state, the pro-inflammatory pathways such as the polyol pathway are activated due to defective insulin signaling, possibly compounded by Aβ oligomers, increased AGE adduct formation, and inflammatory mediators. 59 In this state, the nerve cells are exposed to chronic hyperglycemia, inflammation, oxidative stress, ischemia, and hypoxia, which alter microglial morphology and function across the different regions of the CNS, namely, optic nerve, retina, brain, and spinal cord. 60 Furthermore, this upregulation of inflammatory processes can potentially lead to damage to the BBB endothelium due to the destruction of collagen in the basal lamina surrounding capillaries, and all layers of arteries and arterioles. 61 This damage to the brain’s vasculature may lead to leakage of plasma from the blood vessel into the parenchyma, further glial activation, and a cascade of events leading to further CNS damage. 62
Brain Atrophy
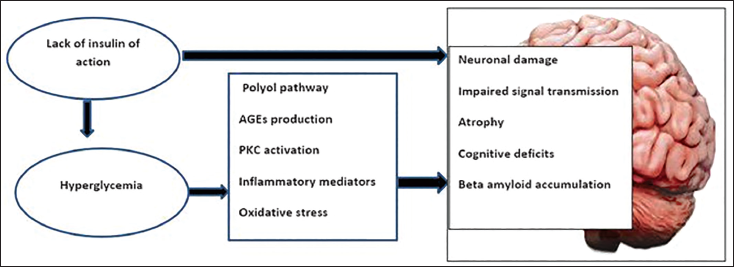
As the disruption in the transmission of signals in the brain continuously progresses, the brain starts to fail to retain new memories. The tangles in the brain nerve cells make it difficult for the cells to communicate, alterations in multiple physiological aspects such as size, shape, and protein expression occur, ACh is reduced, and therefore signal transduction is lost. 63 The profound dysfunction in the CNS discussed above is the key factor resulting in the outward expression of AD symptoms. As the areas in the cerebral cortex responsible for language, reasoning, and social behavior deteriorate, over time, a person with Alzheimer’s gradually loses their ability to live and function independently. 64 Macrovascular damage due to reduced blood flow and synaptic activity of brain cells can result in shrinkage and the observed brain volume loss in Alzheimer’s patients. By promoting microinfarctions, DM results in subclinical, covert mechanisms that lead to brain volume loss. 65 In addition, there has been a strong association between tau and beta-amyloid pathology and cortical atrophy due to the death of neurons. At a macroscopic level, this is reflected in the atrophy of specific brain regions. 66 The huge disparity in brain structure and function between a normal individual and an AD patient is evident. The prospects of AD treatment should investigate optimizing therapeutic targets, including diabetic treatment strategies that can preserve the brain structure and re-establish brain connections. Figure 1 gives an overview and description of the diabetes to AD link as discussed in detail above.
A Diagrammatic Representation of the Impairment of Insulin Activity in the Brain Results in a Defective Insulin Action. In Response to the Hyperglycemic State, There is the Activation of Protein Kinase C, Polyol Pathway, Advance Glycated End Products Formation, Which Favors Inflammation and Reactive Oxygen Species. The Absence of Insulin Action and the Prolonged Pro-inflammatory and Oxidative State Precipitate Neuronal Damage to the Neuronal Components, Leading to Impaired Signal Transmission, Loss of Synaptic Contact, and Brain Atrophy, Ultimately Resulting in Symptoms of Alzheimer’s Disease (AD).37–66
Diabetes Therapy: Repurposing Their Role Towards AD Targets
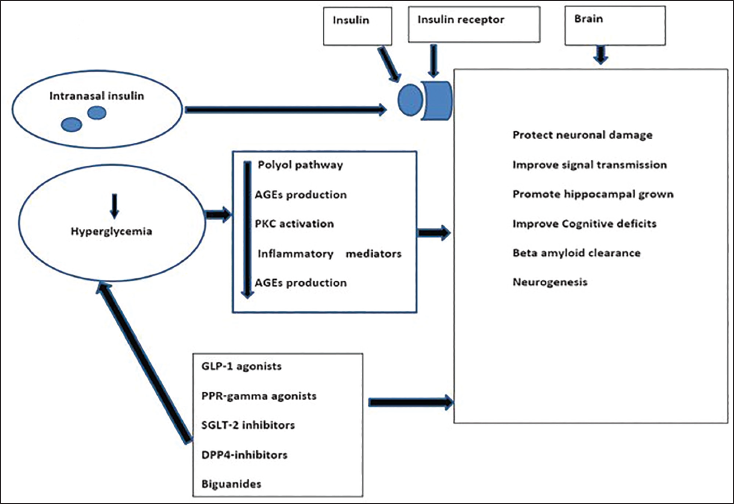
To improve treatment for AD, researchers have looked into targets that aim to nib the dysfunction in processes at the enzymatic and biological levels. In the section discussed above, we have established and synthesized some form of connection between DM and AD. Considering the enormous feat achieved in the treatment of type 2 diabetes mellitus (T2DM), repurposing DM treatment towards AD is a promising area of research due to their pathogenically common features, which include metabolic dysfunction, insulin resistance, and inflammatory processes. This section highlights various diabetes treatments that could be possible molecular targets for AD treatment. Drugs such as glucagon-like peptide-1 (GLP-1) agonists, sodium-glucose co-transporter-2 (SGLT-2) inhibitors, and intranasal insulin delivery are being explored and have shown potential for improving outcomes in AD (Figure 2).
A Simplified Illustration of the Various Anti-diabetic Drugs Investigated for Their Anti-Alzheimer Activity, as Discussed Above. The Brain is the Central Site of Action. The Glucagon-like Peptide-1 (GLP-1), Peroxisome Proliferator-activated Receptor (PPAR)-gamma, Sodium-glucose Co-transporter-2 (SGLT-2) Inhibitors, Dipeptidyl Peptidase-4 (DPP-4) Inhibitors, Biguanides, As Well As Intranasal Insulin Formulation, Both Act on the Hippocampus to Achieve Their Neurological Effect and Increase the Clearance of Beta-amyloid Oligomers, Which Are Known to Result in the Formation of Neurofibrillary Tangles in the Brain. The Overall Effect of These Drugs Would Increase the Cognitive Effect.77–100
GLP-1 Agonist
A class of medication used in T2DM as well as obesity. An example of drugs in this class is exenatide. The hormone GLP-1 exerts its anti-hyperglycemic effects by increasing insulin secretion, decreasing glucagon secretion, delaying gastric emptying, increasing satiety and insulin sensitivity, and including neuro-protection. 67 In a study done on intracerebroventricular streptozotocin (ICV-STZ) treated rat models of AD, exenatide was proposed to suppress the inflammatory response, protect neurons against apoptosis by regulating several autophagic markers, and significantly preserve brain choline acetyltransferase activity, and this neurotransmitter promotes signal transmission between nerve cells, increasing the viability of the hippocampal neuron, and improving cognitive impairment status. 68 In transgenic mice, exenatide prevented impaired insulin signaling in the hippocampus and enhanced cognition, as demonstrated by a decline in hippocampal serine phosphorylation of IRS-1 (IRS-1pSer) and activation of c-Jun N-terminal kinase (JNK). 69 Maintaining neural network integrity and signal transmission between neurons is necessary for brain activities and the effective relay of nerve signals. Therefore, making this drug a target is a potentially viable option for AD treatment.
SGLT2 Inhibitors
In the treatment of diabetes, SGLT2 inhibitors work by preventing the kidneys from reabsorbing glucose, as well as increasing glucose excretion via the urine, which lowers blood glucose concentration. 70 SGLT2 inhibitors have been linked to a reduction in brain oxidative stress and an increase in brain-derived neurotrophic factors. Dapagliflozin, for example, has been proposed to have AChE inhibitory activity, which could be beneficial in the treatment of AD. 71 Dapagliflozin markedly alleviated neuronal oxidative stress and neuroinflammation via lowering lipid peroxides, inhibiting the activation of the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway, and tumor necrosis factor-alpha (TNF-α) levels. 72
Biguanides
This class of anti-diabetics lowers blood glucose concentration by inhibiting hepatic gluconeogenesis and increasing glucose absorption in peripheral tissues. In addition to its anti-diabetic activity, metformin also has beneficial effects for various CNS disorders, such as Parkinson’s.73, 74 Metformin has been shown to enhance mitochondrial function, reducing oxidative stress, inflammation, and amyloid plaque deposition in the CNS. 75 Studies have reported a significantly lower incidence of dementia with long-term use of metformin in T2D patients. In high glucose-cultured HT22 cells, metformin increased autophagy in a dose-dependent manner and attenuated diabetes-induced tau hyperphosphorylation in vitro and in vivo by enhancing autophagy clearance. 73 A cohort study investigating the link between the termination of metformin use in patients and the incidence of dementia concluded that metformin cessation was associated with increased dementia incidence, with another study proposing metformin to be used as a first-line therapy for diabetes in patients at risk of developing dementia or AD.76, 77
Dipeptidyl Peptidase-4 (DPP-4) Inhibitors
In the treatment of DM, DPP-4 inhibitors inhibit the enzyme DPP-4, which is responsible for the degradation of two key incretin hormones: GLP-1 and glucose-dependent insulinotropic polypeptide (GIP). These hormones regulate glucose homeostasis by stimulating the pancreatic cells (α- and β-cells) and regulating insulin and glucagon secretion with respect to blood glucose concentration.78, 79 On the other hand, these drugs inhibit inflammation and oxidative stress through the GLP-1/GLP-1R signaling pathway, affecting the production and clearance of toxic proteins, thereby improving cognitive function. 80 Aβ oligomers disrupt the insulin pathway in the hippocampus either by binding to the insulin receptor or by overactivation of insulin receptor substrate-1 (IRS-1). Hence, by modulating neuronal insulin resistance via the IRS-1 pathway, linagliptin had shown convincing effects against peripheral insulin resistance attributed to the improvement of incretin concentrations as well as attenuation of Aβ oligomers, phosphorylated-tau, and neuroinflammation in the brain of an amyloid-beta (1-42) peptides-induced AD rat model. 81 This makes this target a potential curative approach for alleviating central insulin resistance and AD-related complications.
Insulin Therapy
Insulin and insulin-like growth factor (IGF-1) have intense effects on the CNS, regulating key processes such as energy homeostasis, neuronal survival, longevity, learning, and memory. The binding of insulin or IGF-1 induces a conformational change of the receptor, leading to its autophosphorylation and activation of various signaling pathways involved in the maintenance of synaptic plasticity and memory consolidation.82, 83 Peripheral insulin administration, commonly used to treat diabetes, may cause hypoglycemia in AD patients who do not have diabetes. Furthermore, this method may be ineffective in AD patients due to impaired insulin transport across the BBB. As a result, research has explored the potential of intranasal insulin delivery as an alternative approach.84, 85 Nasal insulin has been found to cross the BBB and enhance memory and cognition in Alzheimer’s patients.86, 87 Early-stage clinical trials and proof-of-concept studies involving individuals with mild cognitive impairment (MCI) or AD have indicated that both short- and long-term intranasal insulin administration can improve memory, suggesting that brain insulin resistance may play a role in Alzheimer’s, regardless of any associated metabolic dysfunction.88, 89 This highlights the potential importance of nasal insulin for targeted brain delivery rather than its systemic use. Thus, the repurposing of nasal insulin for Alzheimer’s treatment is promising.
Research shows that nasal delivery allows insulin to reach the brain rapidly without impacting blood glucose concentration.90, 91 Recent clinical trials have also supported the effectiveness of nasal insulin in managing cognitive disorders. 92 In a randomized, double-blind trial, intranasal insulin reduced markers associated with Alzheimer’s and white matter hypersensitivity. Insulin was administered twice over 12 months, followed by magnetic resonance imaging (MRI) and cerebrospinal fluid collection via lumbar puncture to measure apolipoprotein E (APOE). 93 A study involving individuals with AD or MCI found that 4 months of intranasal insulin helped preserve cognition, as measured by the Alzheimer’s Disease Assessment Scale–Cognitive Subscale (ADAS-Cog), improved delayed memory, and enhanced caregiver-rated functional abilities. 94 Moreover, some research using intravenous and intranasal insulin has shown that the impact of insulin on memory varies depending on the APOE genotype.84, 85 However, earlier trials reported no cognitive benefits from intranasal insulin, though no adverse effects were observed, suggesting its safety.95, 96 Despite mixed results from clinical trials, ongoing developments suggest that nasal insulin may emerge as a treatment for cognitive disorders. Intranasal insulin has been investigated as a potential treatment for cognitive disorders, particularly AD and MCI. Early studies yielded mixed results regarding its efficacy. For instance, a randomized clinical trial published in 2011 found that intranasal insulin administration stabilized or improved cognition and function in individuals with MCI or AD, without significant adverse effects. 97 Subsequent research has continued to explore intranasal insulin’s therapeutic potential. A 2020 study assessed the safety, efficacy, and feasibility of intranasal insulin for treating individuals with MCI, contributing to the growing body of evidence on this intervention. 94 A meta-analysis published in 2022 reviewed multiple randomized controlled trials and concluded that, while intranasal insulin did not show significant cognitive improvements across all studies, certain subgroups, such as Alzheimer’s patients without the APOE4 allele, exhibited enhanced verbal memory performance. The analysis also confirmed the safety of intranasal insulin, as no significant adverse effects were reported. 98 In summary, while early trials reported mixed results regarding the cognitive benefits of intranasal insulin, they consistently indicated its safety. Ongoing research should aim to clarify its potential role as a treatment for cognitive disorders.
Peroxisome Proliferator-activated Receptor-γ (PPAR-γ) Agonists
PPAR-γ agonists, also known as insulin sensitizers, increase tissue responsiveness to lower insulin concentrations. Recently, growing focus has been placed on the role of PPARγ in neurodegenerative diseases, with numerous PPARγ ligands being suggested as potential therapies for cognitive impairment. 98 Their role in preserving or enhancing cognitive function in AD has been linked to in vitro and animal studies demonstrating promising effects, including reduced inflammation, enhanced Aβ clearance, decreased tau hyperphosphorylation, improved insulin signaling, and improved synaptic plasticity.99, 100
Pioglitazone has been shown to significantly reduce neuroinflammation and cerebral oxidative stress, suggesting a potential neuroprotective role through improvements in inflammation-related neuropathy and insulin signaling pathways. 101 Additionally, Yang et al. found that intragastric administration of pioglitazone improved Aβ42 deposition in the hippocampus of insulin-resistant rats by increasing insulin-degrading enzymes and PPAR-γ expression. The treatment also reduced Aβ accumulation via the AKT/GSK3β signaling pathway, with Aβ levels assessed through hippocampal immunohistochemistry.101, 102 For instance, in one study involving 30 patients with MCI and AD, memory improved, and plasma Aβ42 levels stabilized after 6 months of rosiglitazone treatment. 103 Continuous monitoring is crucial during PPAR-γ agonist treatment. The studies reviewed indicated that early intervention with PPAR-γ agonists benefits patients, and high-dose therapy may yield better clinical outcomes, particularly in reversing cognitive decline. 104
Molecular mechanisms by which PPARγ counters cognitive decline require further investigation, as the limited bioavailability of PPARγ ligands in the brain restricts their use as therapeutic agents.101, 105 As a result, a key challenge is developing PPARγ-targeted compounds with improved CNS permeability, 106 which could be achieved by gaining a deeper understanding of the molecular structure of PPARγ and its regulatory effects on various cellular functions. The findings from both in vitro and in vivo models emphasize the beneficial impact of PPARγ agonists in improving cognitive function in AD and suggest these drugs, along with natural PPARγ ligands, as potential future treatments for cognitive impairment.107, 108 While limited brain bioavailability poses a significant challenge for the use of PPARγ ligands in treating cognitive disorders, ongoing research into dual agonists, selective PPARγ modulators (SPPARγMs), and advanced delivery systems offers promising avenues to overcome these obstacles. Researchers have designed novel compounds that selectively target both PPARδ and PPARγ receptors. For instance, the compound AU9 has been developed to improve behavioral deficits and synaptic plasticity while reducing amyloid-beta levels and inflammation in AD models. This dual activation approach may enhance therapeutic outcomes while minimizing side effects. 109 The development of SPPARγMs aims to fine-tune PPARγ activation, providing therapeutic benefits while reducing adverse effects. These modulators selectively modulate PPARγ activity, potentially improving safety profiles and enhancing brain bioavailability. 110 It is also worth mentioning that employing nanoparticles as delivery vehicles can enhance the transport of PPARγ ligands across the BBB. This approach may improve the bioavailability of these compounds in the brain, thereby increasing their therapeutic efficacy in CNS disorders. 111
Conclusion
In conclusion, AD presents unique challenges due to its complex etiology encompassing impaired glucose metabolism, oxidative stressors, neuronal dysfunction, and cognitive decline. Additionally, the BBB poses a significant hurdle in delivering effective treatments to the brain. The use of glucose-lowering medications is relevant and applicable in the treatment of AD due to the overlapping pathological features of diabetes. While dose titration of these medications is crucial for minimizing the risk of hypoglycemia, it must be approached with caution, as cognitive impairments in AD patients may hinder their ability to detect and respond to hypoglycemic symptoms. To address this, innovative drug delivery systems, such as nanoparticles and liposomes, offer promising strategies to enhance brain-targeted delivery of glucose-regulating agents, thereby improving therapeutic outcomes while reducing the risk of hypoglycemia. Ultimately, a tailored approach that considers both the patient’s cognitive and metabolic needs, alongside advancements in drug delivery technologies, is essential to improve safety and efficacy in the management of hypoglycemia in AD.
It is also important to note that treating elderly patients with cognitive impairments presents several challenges in clinical settings. Polypharmacy increases the risk of adverse drug reactions and requires careful medication management. Diagnosing cognitive impairments is complicated by overlapping symptoms and limited access to specialized assessments. The healthcare system fragmentation leads to disjointed care, highlighting the need for better coordination among multidisciplinary teams. Informed consent can be difficult, requiring the involvement of caregivers and/or legal representatives. Limited accessibility to specialized care, financial constraints, and logistical issues further hinder treatment. Additionally, managing medication side effects requires continuous monitoring, placing a burden on healthcare providers and caregivers. Addressing these challenges requires an integrated, patient-centered approach that combines medical, psychological, and social support to improve care for elderly patients with cognitive impairments.
Repurposing diabetes treatments for AD has the potential to transform care by moving beyond traditional palliative approaches and directly targeting the disease process. This will offer a novel approach to slowing or reversing cognitive decline. However, rigorous clinical trials are essential to ensure the safety and efficacy, and to identify potential side effects of these drugs in AD patients, with personalized treatment variations considered where possible. Conclusively, diabetes-related pathology evidently precipitates AD. Therefore, the unfounded progress made in the development of drug therapies for diabetes could infer that finding a cure for AD might not be far-fetched if more therapies looked to address these interlinked targets.
Footnotes
Abbreviations
Aβ: Amyloid beta; ACh: Acetylcholine; AChE: Acetylcholinesterase; AD: Alzheimer’s disease; AGEs: Advanced glycation end products; AKT: Protein kinase B; AMPA: α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; APOE: Apolipoprotein E; ATP: Adenosine triphosphate; BBB: Blood–brain barrier; Cis: Cholinesterase inhibitors; CNS: Central nervous system; DHAP: Dihydroxyacetone phosphate; DM: Diabetes mellitus; DPP-4: Dipeptidyl peptidase-4; ECM: Extracellular matrix; GA3P: Glyceraldehyde-3-phosphate; GIP: Glucose-dependent insulinotropic polypeptide; GLP-1: Glucagon-like peptide-1; GLUT: Glucose transporter; IGF-1: Insulin-like growth factor; IRS-1: Insulin receptor substrate-1; MCI: Mild cognitive impairment; MMPs: Matrix metalloproteinases; NADPH: Nicotinamide adenine dinucleotide phosphate; NMDA: N-methyl-
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This study did not require ethical approval.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
Not applicable.