
Abstract
Background/Objectives
Complications associated with diabetes are predominantly influenced by the polyol pathway, in which aldose reductase 2 (ALR2) is instrumental in converting surplus glucose into sorbitol, resulting in oxidative stress and cellular injury. Targeting ALR2 with natural inhibitors offers a promising therapeutic approach.
Materials and Methods
This study utilized molecular docking-based virtual screening and screened 182 bioactive compounds from Gymnema sylvestre (GS) against ALR2. The co-crystallized ligand (IDD594) served as a benchmark for comparison. The co-crystallized ligand (IDD594) was utilized as a positive control. Additional molecular descriptors and drug-likeness evaluations were conducted utilizing the local online traditional use system (LOTUS) database.
Results
Among the screened compounds, LTS0010237, LTS0042292, and LTS0072617 demonstrated superior binding affinities compared to the control, engaging with critical ALR2 residues. The analyses demonstrated advantageous lipophilicity, solubility, and structural stability. The top three compounds complied with Lipinski’s Rule of Five (RO5), indicating favorable pharmacokinetic characteristics.
Conclusion
The results underscore the therapeutic potential of compounds derived from GS as ALR2 inhibitors, necessitating further validation to evaluate their effectiveness in alleviating diabetes-related complications.
Introduction
Diabetes mellitus is a progressive metabolic disorder marked by chronic hyperglycemia, resulting in various complications that impact multiple organ systems.1, 2 The polyol pathway is an essential molecular pathway involved in these complications, with aldose reductase 2 (ALR2) playing a significant role in its activation. 3 ALR2 facilitates the transformation of glucose into sorbitol, leading to osmotic imbalance, depletion of nicotinamide adenine dinucleotide phosphate (NADPH), and heightened oxidative stress. This metabolic dysregulation promotes the accumulation of reactive oxygen species (ROS) and advanced glycation end-products (AGEs), thereby accelerating the progression of diabetic neuropathy, nephropathy, and retinopathy. 4 Given its pivotal role in diabetes-associated microvascular damage, targeting ALR2 with selective inhibitors presents a promising approach. Nevertheless, inadequate selectivity for its homolog, ALR1, has impeded the advancement of efficacious ALR2 inhibitors, leading to off-target effects and toxicity issues. 5
Despite the potential advantages of ALR2 aldose reductase inhibitors (ARIs), their clinical development has faced numerous obstacles. Promising preclinical and initial clinical results have emerged from various synthetic ARIs, including sorbinil, fidarestat, tolrestat, and ranirestat.6–8 Epalrestat is currently the only clinically approved ARI, and it is primarily used in Japan and other Asian countries to treat diabetic neuropathy. 9 Numerous studies have explored natural inhibitors of ALR2; however, there remains considerable potential for discovering new bioactive compounds that demonstrate improved selectivity and efficacy in addressing diabetes-related complications.10, 11
Drug development is a multifaceted, sequential process focused on identifying and optimizing small molecules or biomolecules as prospective therapeutics. This extensive and expensive undertaking, lasting 10–15 years with costs totaling $2.56 billion, encompasses target identification, hit discovery, lead optimization, and preclinical and clinical assessments. 12 To improve efficiency and decrease expenses, computer-assisted drug discovery (CADD) has emerged as a crucial instrument, expediting preliminary research and diminishing late-stage failures. 13
Gymnema sylvestre (GS) is widely acknowledged for its glucose-lowering properties and its capacity to regulate carbohydrate metabolism among the numerous medicinal plants with anti-diabetic potential.14, 15 Gymnetic acids, gymnemagenin, flavonoids, and saponins are bioactive compounds in GS that have pharmacological effects, such as modulating insulin secretion, inhibiting glucose absorption, and regenerating pancreatic β-cells. Despite extensive research into GS’s anti-diabetic properties, its potential role as a selective ALR2 inhibitor is underexplored.16, 17 This study performs a computational screening of the bioactive compounds of GS concerning the ALR2 binding sites and their interactions to identify potential natural ALR2 inhibitors.
Materials and Methods
ALR2 Structure Retrieval and Preparation
The 3D structure of ALR2 (PDB ID: 1US0) was sourced from the protein data bank (PDB). 18 The protein underwent preprocessing and refinement using Discovery Studio Visualizer, which eliminated water molecules and heteroatoms, enhancing structural integrity to ensure optimal docking precision. The co-crystallized ligand derived from the structure was retained and employed as a positive control in the comparative docking analysis. In the context of PyRx molecular docking simulations, the preprocessed ALR2 protein underwent conversion to protein data bank, partial charge (Q), and atom type (T) (PDBQT) formats to guarantee its compatibility with AutoDock Vina.
GS Compound Library Preparation
A total of 182 bioactive compounds were retrieved from the local online traditional use system (LOTUS) database. 19 The compounds were acquired in structure data format (SDF) to ensure compatibility with molecular docking tools and were subsequently transformed to PDBQT using Open Babel. Furthermore, PyRx used the Merck molecular force field 94 (MMFF94) force field for energy minimization, iteratively optimizing atomic coordinates to reduce overall potential energy, resulting in a low-energy, reproducible conformation for improved docking accuracy. 20
Structure-based Virtual Screening
The PyRx tool was employed for structure-based virtual screening to evaluate the binding interactions between ALR2 and phytochemicals from GS. The co-crystallized ligand (IDD594) served as a reference ligand for identifying the active site of ALR2. The grid box was positioned at X: 16.47, Y: –7.21, and Z: 15.07, facilitating accurate docking within the binding pocket. The docking results were examined to determine the phytochemicals’ critical interaction residues and binding affinities to ALR2. Compounds exhibiting elevated binding affinities, advantageous molecular interactions, and stable docking conformations were chosen for subsequent evaluation and absorption, distribution, metabolism, excretion, and toxicity (ADMET) analysis.
Molecular Properties and Descriptors Analysis
The molecular properties and descriptors for the three compounds of GS were acquired from the LOTUS database. The molecular properties and descriptors were extracted directly from the LOTUS database records. The molecular properties extracted were total atom, heavy atom, bond count, number of carbons, minimum number of rings, and maximum number of rings. Furthermore, the molecular descriptors analyzed included the natural product (NP)-likeness score, Alogp, Alogp2, Apol, Bpol, Lipinski’s Rule of Five (RO5), Wiener Path Noo, Xlogp, Zagreb Index, and TopoPSA, ensuring detailed and reproducible compound characterization.
Results
This study aimed to identify potential ALR2 inhibitors from natural compounds in GS through molecular docking, virtual screening, and ADMET analysis. The screening effectively identified promising inhibitors that exhibited improved binding specificity and stability compared to compounds that had been previously examined. A total of 182 bioactive compounds were docked to the active site residues of ALR2, and their interactions were compared with the co-crystallized positive control (IDD594) to evaluate their inhibitory potential and binding efficacy. IDD594 is a potent inhibitor of ALR2, an enzyme linked to diabetic complications such as neuropathy, retinopathy, and nephropathy. Its binding mechanism and efficacy have been extensively studied. 21
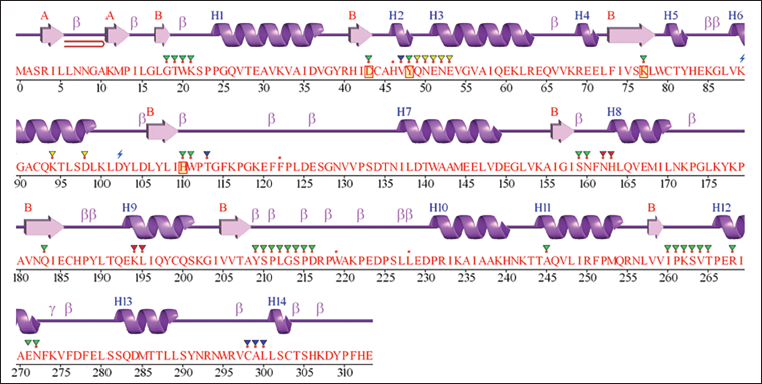
First, the secondary structure of ALR2 was analyzed with ProMotif, revealing a well-organized structural framework required for enzymatic function and ligand interactions. The protein has two β-sheets, 10 β-strands, and seven β–α–β units that make up its core β-sheet structure. The presence of one β-hairpin and one β-bulge helps stabilize the β-sheet arrangements. The protein’s 14 α-helices help maintain structural integrity and facilitate substrate binding. Eleven helix–helix interactions support the protein’s stability, while 25 β-turns and one γ-turn allow for conformational changes during ligand binding. These structural features are essential in understanding the binding interactions of ALR2 inhibitors because they affect the accessibility and specificity of the active site, guiding the rational design of selective inhibitors for diabetic complications (Figure 1).
The Secondary Structure of Aldose Reductase 2 (ALR2) (PDB ID: 1US0), Displays β-strands (Arrows), α-Helices (Coils), β-turns, and γ-turns, Highlighting the Structural Organization of the Protein.
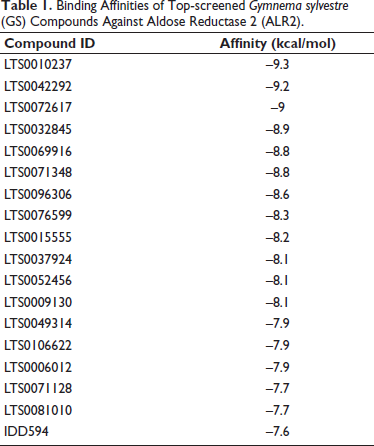
The prepared library of bioactive compounds and the target protein (ALR2) underwent virtual screening via PyRx, utilizing IDD594 as the positive control ligand. Of the 182 screened molecules, 18 compounds displayed greater binding affinity than the control, indicating enhanced interactions with the ALR2 active site residues (Table 1).
Binding Affinities of Top-screened Gymnema sylvestre (GS) Compounds Against Aldose Reductase 2 (ALR2).
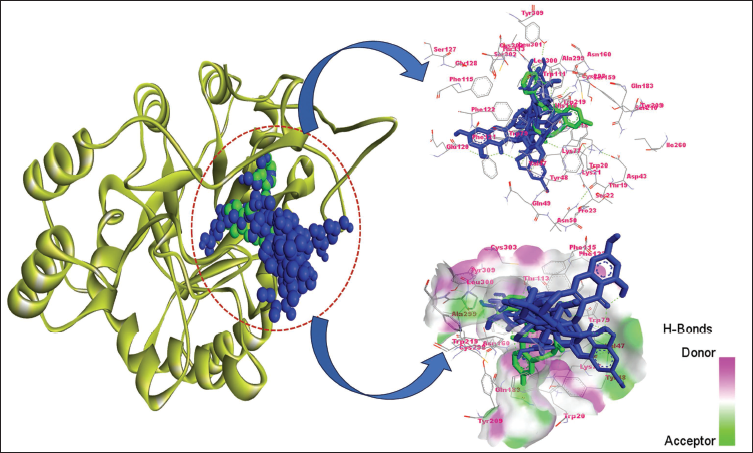
The molecular docking analysis revealed that the top three screened compounds (LTS0010237, LTS0042292, and LTS0072617) had strong binding affinities for ALR2, comparable to or better than the control. The detailed two-dimensional (2D) and three-dimensional (3D) interaction analysis (Figure 2) revealed that these compounds effectively bind to the ALR2 active site, engaging key residues in interacting with the control ligand. The screened ligands’ overlapping interactions with common ALR2 residues indicate a high potential for selective ALR2 inhibition (Figure 2).
Molecular Interactions of the Top Three Compounds With Aldose Reductase 2 (ALR2). The Control Ligand (IDD594) is Green, While the Top Three Screened Compounds (LTS0010237, LTS0042292, and LTS0072617) are Blue. The Left Panel Highlights the Overall Binding Site, While the Top-right and Bottom-right Panels Depict 2D and 3D Interaction Maps, Showing Key ALR2 Residues Involved in Ligand Binding. Hydrogen Bond Donors and Acceptors are Marked in Pink and Green, Respectively.
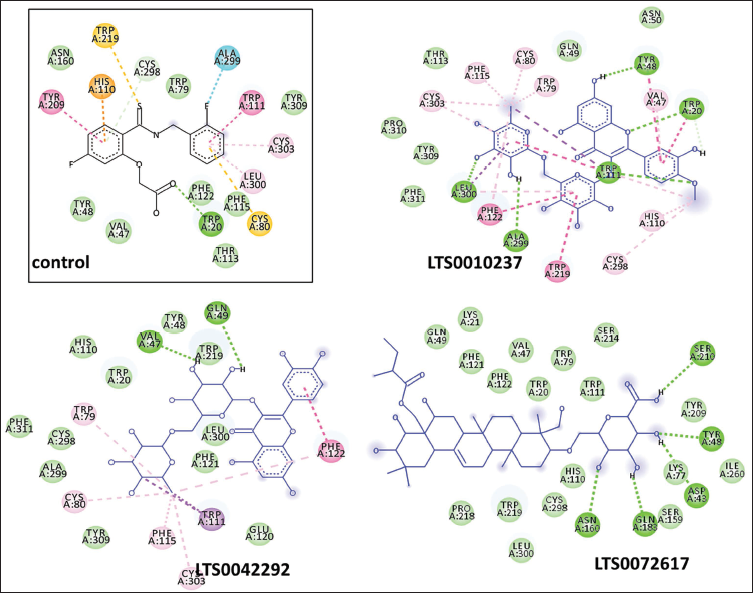
A 2D interaction analysis was performed with Discovery Studio Visualizer to understand the binding interactions better, comparing the top three screened compounds to the control (Figure 3). The control ligand formed multiple hydrogen bonds and hydrophobic interactions with key ALR2 residues, such as His110, Trp219, Cys298, Phe122, and Leu300, all of which are required for enzyme inhibition. Similarly, the three selected compounds had strong binding interactions with the ALR2 active site, with binding affinities comparable to or superior to the control ligand.
LTS0010237 formed hydrogen bonds and hydrophobic interactions with Leu300, Trp219, and Phe122, indicating a stable interaction. LTS0042292 formed multiple hydrogen bonds with residues, including Trp20, His110, and Phe122, indicating a high binding affinity and complementarity within the active site. Meanwhile, LTS0072617 exhibited extensive interactions with ALR2, forming hydrogen bonds with Ser210, Asn160, and Tyr209, which increased its stability within the binding pocket. These compounds’ overlapping interactions with the critical active site residues indicate their potential as selective ALR2 inhibitors.
Two-dimensional (2D) Interaction Analysis of the Top Three Compounds with Aldose Reductase 2 (ALR2).
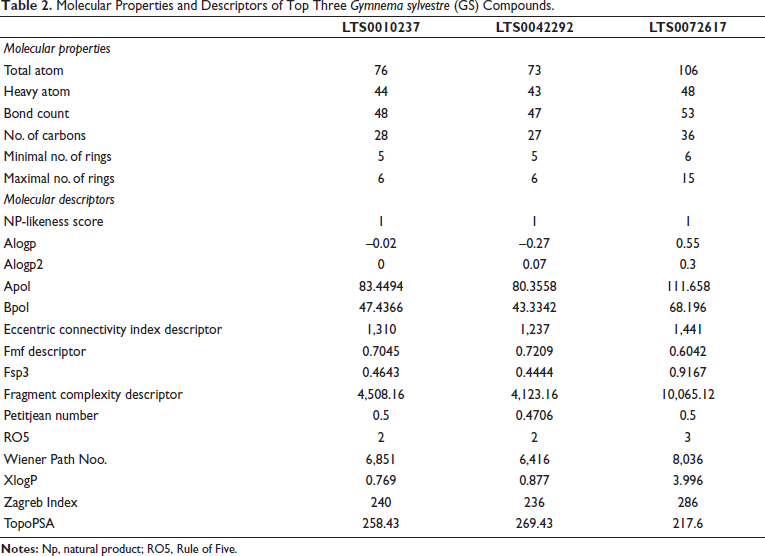
The top three ALR2-binding compounds from GS were examined for molecular properties and descriptors, revealing structural stability, drug-likeness, and potential inhibitory activity. All compounds exhibited moderate to high molecular complexity, with 5–15 rings forming a stable core framework for ALR2 binding.
The NP-likeness score of one confirmed their natural origin, while AlogP and XlogP values indicated varying lipophilicity, with LTS0072617 having the highest membrane permeability (XlogP = 3.996). Strong Apol and Bpol values indicate favorable polar interactions required for hydrogen bonding within the ALR2 active site. Lipinski’s RO5 analysis confirmed the drug-like properties. LTS0072617 had a higher molecular and fragment complexity (10,065.12), indicating more possible protein-ligand interactions. TopoPSA values ranged from 217.6 to 269.43, indicating good solubility and bioavailability (Table 2).
Molecular Properties and Descriptors of Top Three Gymnema sylvestre (GS) Compounds.
Discussion
The molecular docking analysis revealed the promising potential for natural ALR2 inhibitors from GS, with three compounds (LTS0010237, LTS0042292, and LTS0072617) demonstrating superior binding affinities compared to the control ligand IDD594. These compounds exhibited strong interactions with crucial ALR2 residues, including His110, Trp219, Cys298, Phe122, and Leu300, which are essential for enzyme inhibition. The formation of multiple hydrogen bonds and hydrophobic interactions suggests stable binding conformations, particularly noteworthy in LTS0072617, which showed extensive interactions with Ser210, Asn160, and Tyr209. These interaction patterns indicate that these compounds could potentially achieve selective ALR2 inhibition while maintaining binding stability comparable to established inhibitors.
The molecular properties and descriptors analysis further supports the therapeutic potential of these compounds. All three lead compounds demonstrated favorable drug-like characteristics, adhering to Lipinski’s RO5, which suggests good oral bioavailability. Particularly interesting is LTS0072617’s higher molecular and fragment complexity (10,065.12), indicating more potential protein-ligand interactions, while its optimal TopoPSA value (217.6) suggests good solubility and bioavailability. The balanced lipophilicity values, especially LTS0072617’s XlogP of 3.996, indicate appropriate membrane permeability without compromising solubility. These physicochemical properties, combined with the strong binding affinities, suggest that these compounds could serve as promising scaffolds for developing natural ALR2 inhibitors with potentially fewer side effects than synthetic alternatives.
Conclusion
This study computationally identified potential ALR2 inhibitors from GS through molecular docking-based virtual screening, ADMET analysis, and evaluation of molecular descriptors. Of the 182 screened compounds, LTS0010237, LTS0042292, and LTS0072617 exhibited robust binding affinities, advantageous drug-like characteristics, and structural stability akin to the control. The findings underscore the therapeutic potential of these bioactive compounds and necessitate further validation for their advancement as natural ALR2 inhibitors for diabetes-related complications.
Footnotes
Abbreviations
2D: Two dimensional; 3D: Three dimensional; ADMET: Absorption, distribution, metabolism, excretion, and toxicity; AGEs: Advanced glycation end-products; ALR2: Aldose reductase 2; ARIs: Aldose reductase inhibitors; CADD: Computer-assisted drug discovery; GS: Gymnema sylvestre; LOTUS: Local online traditional use system; MMFF94: Merck molecular force field 94; NA: Not applicable; NADPH: Nicotinamide adenine dinucleotide phosphate; PDB: Protein data bank; PDBQT: Protein data bank, partial charge (Q), and atom type (T); ROS: Reactive oxygen species; SDF: Structure data format; TopoPSA: Topological polar surface area.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval and Informed Consent
Not applicable.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research has been funded by the Scientific Research Deanship at the University of Hail, Saudi Arabia, through project number RG-21 162.