
Abstract
This review article explores the relationship between alpha-synuclein and mitochondrial dysfunction in Parkinson’s disease (PD), focusing on the role of hypoxia as an intermediate factor. The interaction between alpha-synuclein and mitochondria, particularly through membranal lipids such as cardiolipins, is highlighted as a key factor in mitochondrial disruption and neurodegeneration. Hypoxia, caused by oxygen deprivation, is identified as a crucial link between alpha-synuclein and mitochondrial regulation, leading to neuronal death in PD. The article also discusses the involvement of other proteins, such as peroxisome proliferator-activated receptor gamma coactivator, Sirtuin-1, Sirtuin-3 and adenosine monophosphate-activated protein kinase, in maintaining mitochondrial biogenesis during hypoxia. The study emphasizes the need for further research to understand the complex molecular interactions causing Lewy body aggregation, improper mitochondrial functioning and neurodegeneration in PD, with a specific focus on the role of hypoxia. Alpha-synuclein aggregation disrupts mitochondrial respiration, leading to mitochondrial dysfunction and increased production of reactive oxygen species. Mitochondrial dysfunction, in turn, causes neurodegeneration in PD. Oligomeric alpha-synuclein results in mitochondrial dysfunction, lethal synaptic disruption and reduced adenosine triphosphate generation. Oligomeric alpha-synuclein also increases the accumulation of mitochondrial rho nucleotide guanosine triphosphate, leading to delayed mitophagy. Hypoxia, another factor in PD, alters both alpha-synuclein and mitochondria. Controlling hypoxia reduces the oligomerization of alpha-synuclein. The interaction between alpha-synuclein and mitochondria is complex, and determining the primary player in inducing the other is still debatable.
Introduction
Parkinson’s disease (PD) is characterized by the loss of dopaminergic neurons in the substantia nigra associated with motor and non-motor symptoms. The pathology for PD is still vague, although a major factor is associated with Lewy bodies and their role in mitochondrial biogenesis.1, 2 The role of monomeric alpha-synuclein and mitochondria thoroughly maintains the neurons, as the oligomeric form of alpha-synuclein disrupts various metabolic functions of mitochondria such as the electron transport chain as it is also involved in adenosine triphosphate (ATP). 3 Hypoxia plays a key role in the formation of neurological disorders, impacting brain ageing and mortality. Mitochondria use much of the oxygen supplied to the body to produce ATP via the electron transport complex mechanism. Hypoxia is also indulged in activating mitophagy which regulates the removal of unwanted mitochondria required to maintain quality control as mitophagy also induces inflammation and increases ROS and nitrosative agents.4–6 It is necessary to conduct a thorough investigation into the molecular mechanism by which hypoxia heightens oxidative and nitrosative stress, which in turn triggers alpha-synuclein aggregation, generates reactive oxygen species and ultimately results in neurodegeneration. Multiple system atrophy in the brains of PD patients is associated with hypoxic factors such as chronic hypoxia, hence, it is evident that oligomeric alpha-synucleins play a major role in the dysfunctioning of mitochondria. 7 The reason for the hypoxia may cause alpha-synucleins to migrate into the cytosol of the mitochondria, although it is still unknown how they migrate across the surface of the mitochondria. 8 Alpha-synuclein regulates mitochondrial dynamics such as fusion and the removal of toxic components. 9 Alpha-synuclein could also be a bystander in producing neurodegeneration. In this review article, we propose various non-highlighted routes for the connection between alpha-synuclein and mitochondria by the mediating term hypoxia, which is involved in neuronal death in PD.
Defective Mitochondria and alpha-synuclein Role in PD
On the ground level, we can assume two main correlated pathological proceedings that have an interest in understanding PD development alpha-synuclein oligomer accumulation, and improper mitochondria biogenesis. Multiple system atrophy is an intensifying neuron disease caused by the oligomeric form of alpha-synuclein at oligodendrocytes in glial cytoplasmic inclusions. 10 Some pathological factors that have been vaguely linked to PD include mitochondrial dysfunction. A rise in oxidative stress and neuroinflammation activates microglial cells, leading to the activation of inflammatory mediators. 11 The mutually stimulating and antagonist interactions among the mentioned factors show challenging research in PD. A thorough understanding of the exchange of involved pathways is necessary to adapt PD models for obtaining successful treatments. This article focuses on the molecular pathway interactions that lead to LB aggregation, improper mitochondrial functioning and neurodegeneration, with a particular emphasis on hypoxia as a mid-key player in these processes. However, rising documentation of disease pathology determines the effect of hypoxia correlating mitochondrial proteostasis in PD. We must focus on a unified model, which should stimulate new research correlating hypoxia in PD which will generate effective PD models.
Alpha-synuclein and PD Focus
Owing to the alpha-synuclein (Lewy bodies) aggregation, alpha-synuclein is hypothesized to be the main cause of PD. 12 The primary mechanism in the formation of LB is the cross-breeding binding interaction between alpha-synuclein and other proteins, such as tau-protein and beta-amyloid. Tau proteins are involved in the formation of neurofibrillary tangles inside neurons, resulting in neuronal death, whereas beta-amyloid accumulation can upregulate the accumulation of tau-protein, again a pathology which is involved in the neuronal damage in Alzheimer’s disease; the tau-protein and beta-amyloid have also been involved with the Lewy body disorders (alpha-synuclein). Research has indicated that in the advanced stages of Lewy body disorders, there is a co-occurrence of neocortical disorders via alpha-synuclein, tau and amyloid. The relation between serum and cerebrospinal fluid levels of tau-protein and beta-amyloid was found in the blood markers of patients with PD. 13 According to a hypothesis, there is growing evidence that LB is present in SN and that it is also stimulated in brainstem nuclei and the dorsal motor nucleus of the vagal nerve in the medulla. There has been research on the potential connection between hypoxia and nitrogen-containing groups and ROS, which leads to mitochondrial dysfunction and a variety of neuronal disorders, including PD. Nevertheless, an increased buildup of alpha-synuclein in the SN region of the brain has indicated a faster rate of PD progression. When alpha-synuclein is overexpressed in synucleinopathies-related neural damage, there is an increased need for oxygen and bioenergy, which results in the death of dopaminergic neurons. 14
Improper Mitochondrial Biogenesis in PD Gaps in the Powerhouse
The involvement of mitochondrial dysfunction is a key factor in the pathology of PD. 15 Langston et al. discovered the accidental interaction of toxic 1-methyl-4-phenyl-1,2,3,6-tetrahydrodropyridine that neutralized the complex-1 electron transport chain. 16 MPTP is used to produce neurotoxicity by causing mitochondrial-based cell apoptosis, oxidative stress and inflammation, resulting in dopaminergic neurodegeneration. 17 1-methyl-4-phenylpyridinium-induced models have shown how upregulation and downregulation of monoamine oxidase can prevent dopaminergic neurodegeneration. 18 MPTP metabolite, MPP+, inhibits complex I of the mitochondrial electron transport chain resulting in ATP reduction and neuronal death. 19 Most complex I inhibitors such as rotenone develop PD symptoms with dopaminergic neuronal death, although most complex I inhibitors mainly do not cause dopaminergic neuronal death as their major target is mitochondrial ATP production, not the cell death pathways that are specific to dopaminergic neurons. 20 Mitochondria are involved in many cycles, including those involving ATP synthesis, lipid synthesis, ROS, nitrogen species balance, apoptosis and Ca2+ homeostasis, which are critical for neuronal survival. Genetically mutated mice models in PD have shown aggregation of alpha-synuclein. 21 Mitochondrial dynamics failure has shown a relation with neuropathies and neuronal death as it regulates biogenesis and degradation in cells. Interestingly, PD is also exhibited by mitochondrial impairment, primarily linked to alpha-synuclein mentioned in the previous studies.22–23 Alpha-synuclein enters mitochondria and inhibits complex I of the electron transport chain.
Alpha-synuclein and Lipid Membrane Interactions
Although alpha-synuclein’s relationship to lipids has been explained in several studies, including how it is linked to oxidized lipids and may cause mitochondrial dysfunction, it is still unknown how lipid membranes contribute to the enhancement of alpha-synuclein aggregation. Numerous recent studies demonstrated how polyunsaturated fatty-acid chains improve the interaction between alpha-synuclein and the membrane. 24 Alpha-synuclein’s N-terminal site’s interactions with phospholipids have been partially explained through in vitro studies, but more investigation is needed to comprehend these interactions with in vivo studies. 25 Alpha-synuclein mostly interacts with negatively charged lipids and forms tubules triggering aggregation. Regarding the presence of lipids and alpha-synuclein in Lewy bodies, there is a lack of clarity. Here, with permeability factors in mind, 50% of the lipid (cholesterol) in the plasma membrane is found in the inner monolayer of synaptic vesicles, where excess lipid is present and demonstrates the interaction between alpha-synuclein and plasma membrane-producing neurodegeneration. 26 Alpha-synuclein and protein interaction in the synapses has been shown to produce toxicity in the synaptic end regions causing neurodegeneration. Numerous alpha-synuclein-related PD model investigations have identified membrane-based trafficking pathways, which demonstrate the capacity of alpha-synuclein aggregation through cellular penetration. 27 Excessively accumulated ganglioside (lipid), in neurons, impacts the alpha-synuclein confirmational kinetics. In a dynamic study using calcium ion with monomeric alpha-synuclein in identifying the confirmational changes, the N-terminal (Figure 1) was exposed which leads to more aggregation of alpha-synuclein. 28 Oligomerization studies have shown how small oligomers produce alpha-synuclein neurotoxicity, and ganglioside model membranes consisting of rich plasma membrane microdomains illustrate how it acts as a moderator alpha-synuclein. 29
N-Terminals From 1 to 60 Strands Consisting of +4 Charge Carrying Mutational Regions A30P, E46K, H5OQ, G51D and A53T in Alpha-synuclein. N-Terminals from 61 to 95 strands consist of the hydrophobic region with –1 charge with the non-amyloid component. The C-terminal with 96–140 carries –12 charge consisting of Serine129 regions that get phosphorylated into Lewy bodies.
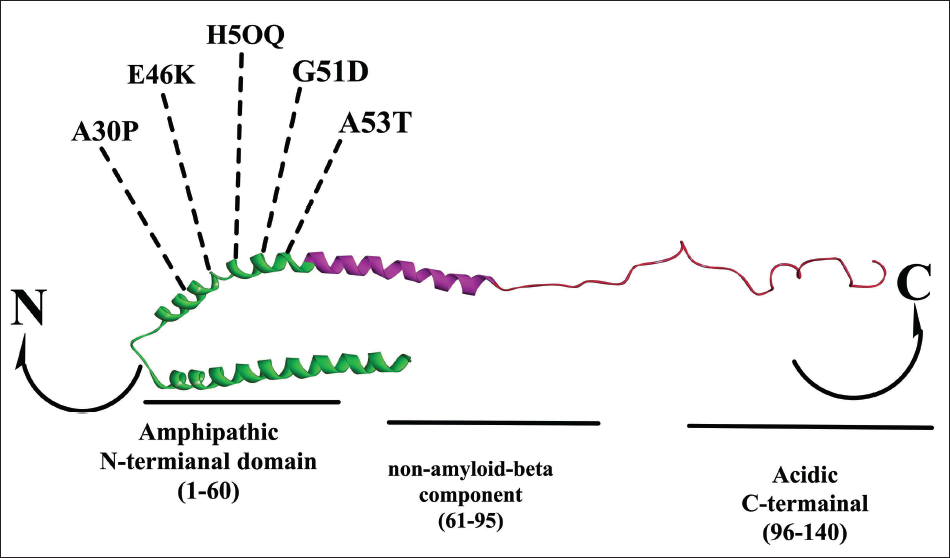
This may help us to understand the pathophysiology of alpha-synuclein aggregation by emphasizing the central key role of lipids and opening a new therapeutic avenue to stop additional neurodegeneration. Figures 2 and 3 show how the transition of accumulated beta-sheet conformation plays a crucial role in the development of LB, which forms protofilament and mature fibrils.
Alpha-synuclein Binding in Various Mitochondrial Membranes, IMM and OMM.
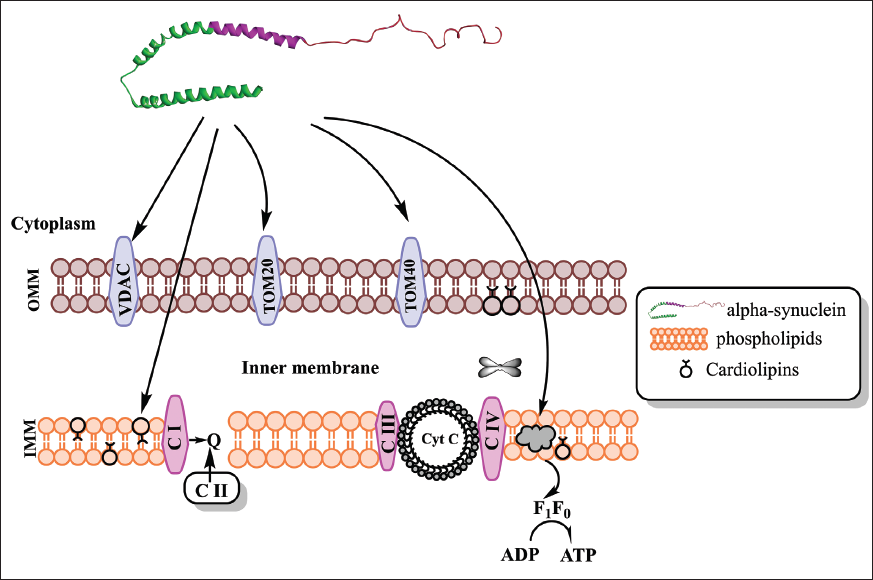
Alpha-synuclein Considering an Intrinsically Disordered Protein. Monomer alpha-synuclein in the dynamic phase gets bound to multimer confirmation in synaptic vesicles.
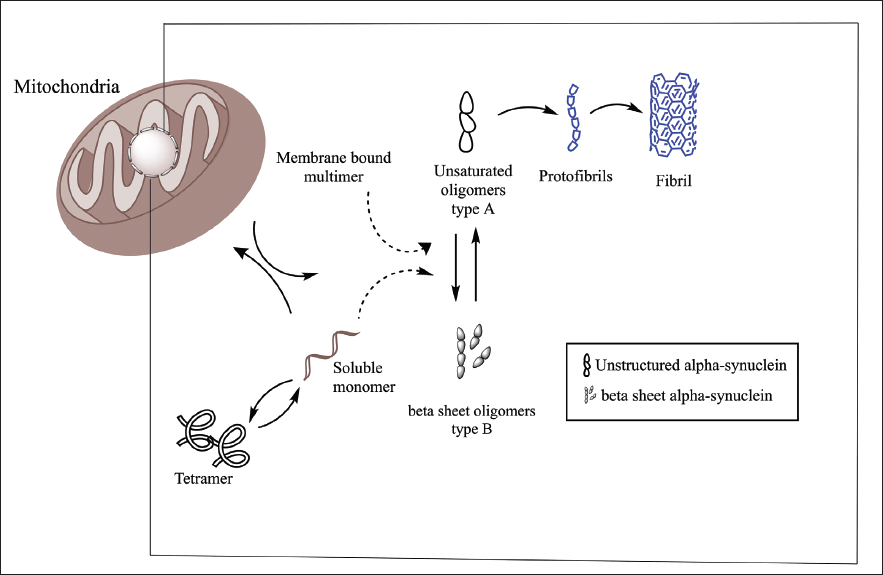
Linkage of Mitochondria and alpha-synuclein
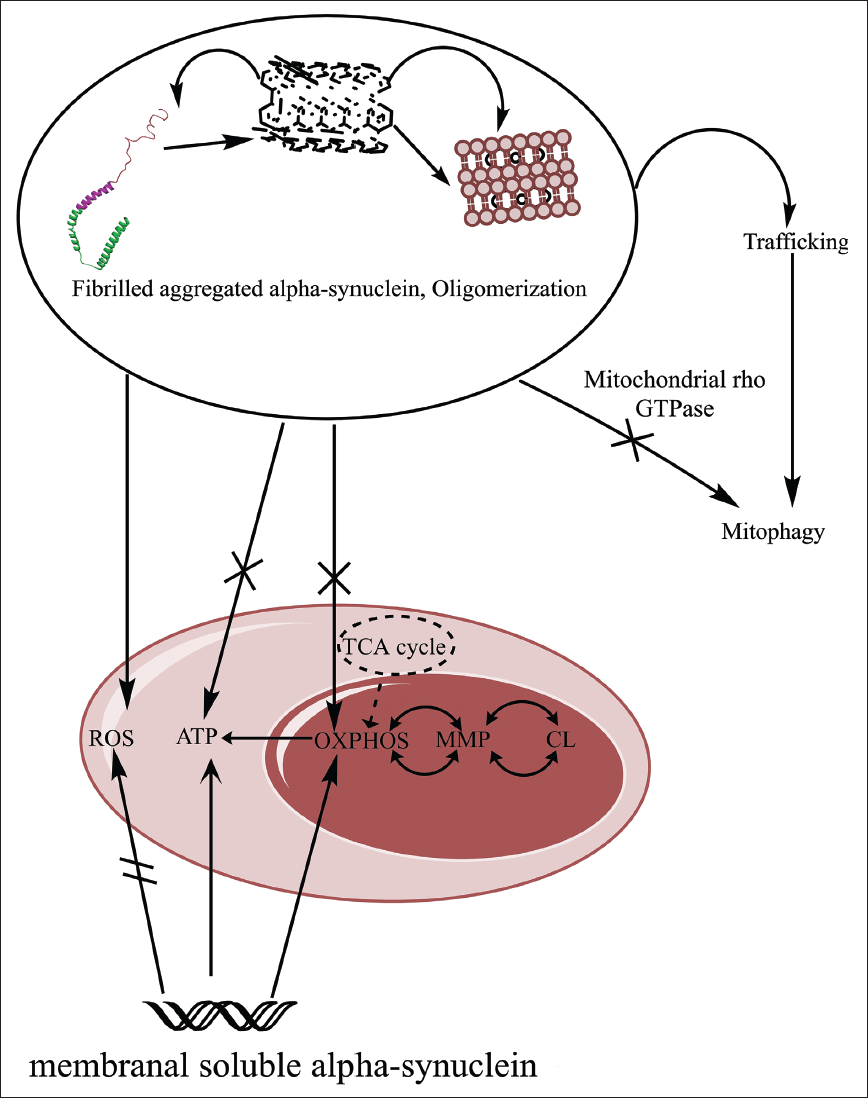
Mitochondrial biogenesis is important in the maintenance of healthy neurons. However during the disruption of complex I–IV of the electron transport chain, there is the leakage of electrons, resulting in ROS and oxidative stress. Alpha-synuclein is also involved in the generation of ROS and oxidative stress. Both in vitro and in vivo models have partially explained alpha-synuclein toxicity in mitochondria. The N-terminal of alpha-synuclein explains the linkage of lipids with functional and pathological factors of PD. Acetylation of N-terminal alpha-synuclein (post-translational modification) has provided its affinity to bind towards lipid membranes; the acetylation of the N-terminal increases alpha-synuclein helical folding property, and this modification can result in over-accumulation of alpha-synuclein, which may result in neurotoxicity. 30 The N-terminal and C-terminal help in increasing fibril aggregation more rapidly. Mutations in the N-terminal of A53T could be the probable cause of alpha-synuclein toxicity resulting in mitochondrial dysfunctioning. In vivo models have shown dopaminergic neurotoxicity via point mutations in alpha-synuclein, fabricating the 71–82 amino acids of alpha-synuclein resulted in no aggregations or oligomerization in the Drosophila model, whereas the trim form of alpha-synuclein resulted in the major formation of aggregations, which suggest that alpha-synuclein has a major role in the neurotoxicity of dopaminergic neurons. The N-terminal domain of the alpha-synuclein is involved with the electron transport chain resulting in mitochondrial elongation; the translational formation of alpha-synuclein to mature fibrils results in oligomers, where they interact with TOM20 and disrupt the mitochondria. This process downregulates the process of mitochondrial respiration resulting in more ROS production. TOM20 binds with oligomeric alpha-synuclein instead of receptor TOM22 and results in neuronal death. The probable route of alpha-synuclein entering into the mitochondria is still unclear, but the consequences of the oligomeric form of alpha-synuclein in the mitochondria result in mitochondrial failure. Alpha-synuclein’s oligomeric forms cause both ATP depletion and disruption of the IMM. The function of alpha-synuclein in the ATP synthase pathway has been explained by several experimental findings on SH-SY5Y cell lines. However, the oligomeric form has downregulated ATP synthesis efficiency in mitochondria, which suggests that a more extensive study between the alpha-synuclein and complex I could result in the findings of more unexplored pathways. 31 A critical study between alpha-synuclein and mitochondrial proteins, and various in vivo models, should be approached. Hypoxia could be the other factors that may be altering both alpha-synuclein and mitochondria. A study on the δ-opioid receptor that regulates alpha-synuclein under hypoxia or MPP+ showed how controlling hypoxia reduces oligomerization of alpha-synuclein (Figure 4). 32 As OXPHOS is triggered during the electron transport chain that occurs on then IMM which results in ATP production, mitochondrial membrane potential (MPP) is involved in the electrochemical gradients (movement of ions through the membranes) that are generated during the electron transport chain activity, and CL generally interacts with the proteins that are involved in oxidative phosphorylation, mitochondrial dynamics and apoptosis; the interaction between OXPHOS, MPP and CL are interconnected mechanisms that are important in maintaining mitochondrial structure, function and integrity, as shown in Figure 5.
Factors Getting Upregulated and Downregulated Due to the Interaction of Alpha-synuclein and Mitochondria.
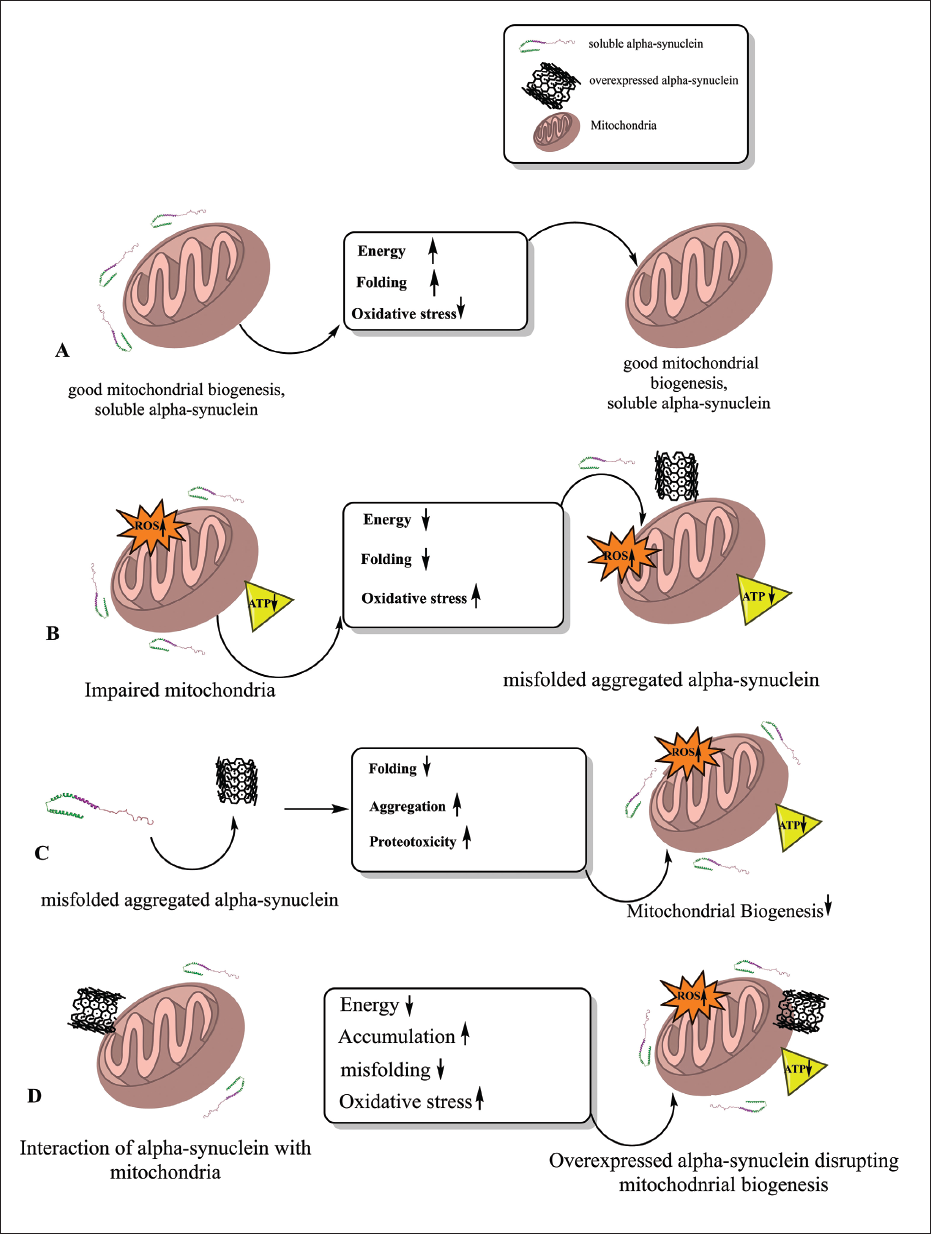
Synaptic Alpha-synuclein Playing a Major Key Role in the Pathology of PD via Disrupting Mitochondrial Functioning. A single unit of alpha-synuclein mainly regulates mitochondrial activities, but an oligomeric unit of fibril alpha-synuclein results in mitochondrial dysfunctioning and lethal synaptic disruption. OXPHOS occurring during the electron transport chain in mitochondria may result in ROS. The TCA cycle involved in regulating the complex I transport chain is disrupted and the generation of ATP is reduced due to oligomeric alpha-synuclein. Oligomeric alpha-synuclein increases mitochondrial rho nucleotide guanosine triphosphate that is over-accumulated in OMM and delays mitophagy.
Cardiolipins and Mitochondria
CL is a phospholipid, located in the IMM and synthesized by phosphatidylglycerol and cytidine-diphosphate-diacylglycerol. Mitochondrial CL consists of glycerol and acyl chains, which play an important role in the stability of enzymes involved in metabolism and energy production. Although CL is produced in the IMM, it is also observed in the OIM, where the surfacing of the CL mostly happens during the mitochondrial injury. 33 As CL is made up of four acyl groups, it is also involved in mitochondrial activities including the electron transport chain, apoptosis and mitophagy. Cytochrome-c and CL work together to reduce ROS and oxidative stress; post-translational modification of cytochrome-c reduces mitochondrial membrane potential and electron transport flux, which minimizes the generation of ROS production and oxidative stress. Cytochrome-c is also linked with lipids, which leads to the oxidation of CL resulting in cell apoptosis. Alpha-synuclein and the formation of mitochondrial pores have both been linked to CL; in alpha-synuclein knockout mice, a decreased level of CL in the mitochondria suggested deficiencies in mitochondrial bioenergetics, as CL is present on the outer surface of mitochondria. They help in the aggregation of alpha-synuclein. It is still unknown how CL moves from the IMM to the OIM and functions as a buffer for the oligomeric alpha-synuclein, but the oligomeric alpha-synuclein aids in the opening of mitochondrial pores through cytochrome-c leakage, which causes the mitochondria to swell and eventually causes apoptosis, where this interaction could be highlighted to regulate mitochondrial functioning. CL undergoing peroxidation can also undergo fatty-acid replacement with four oxidized groups in the IMM. In contrast, these oxidation replacements have been also fabricating CL-dependent signalling processes like electron transport. 34 Lipids on oxidation undergo various dysregulated pathways that mainly affect mitochondrial functioning, as brain-specific fatty-acid protein 3 increases alpha-synuclein aggregation. The N-terminal of alpha-synuclein is a key factor in binding with. Various translocase outer membrane TOM20 and translocase inner membrane TOM40 interact with mitochondrial targeting sequences, which suggests that expressing these could reduce alpha-synuclein aggregation; DAC along with TOM20 and TOM40 plays a significant role with cardiolipins (CL), TOM20 and TOM40 bind with alpha-synuclein and stop mitochondrial protein import and disables complex I process, which generated more ROS species and results in mitochondrial dysfunctioning. VDAC is involved in the mitochondrial biogenesis as well as apoptosis. The improper interaction between the cardiolipin and VDAC could result in mitochondrial failure or cell death. 35 Emphasizing CL plays a serious role in terms of alpha-synuclein and its efficacy in mitochondria and its role in brain homeostasis. The question remains “What is the biosynthetic mechanism and its function in the brain”. We could also draw attention to the main scepticism regarding CL’s potential as a biomarker in neurodegenerative illnesses. However, the routes still need to be more organized.
Hypoxia, alpha-synuclein and Mitochondria Future Aspects
Hypoxia could be generally participating between alpha-synuclein and mitochondrial dysfunctioning via different processes; hypoxic stress activates the HIFs that are involved in the maintenance of cellular response. This condition can imitate various molecular mechanism such as oxidative stress and protein degradation impairment, whereas the chronic hypoxic stress can lead to alpha-synuclein phosphorylation at serine 129 and result in LB aggregation. This aggregation can lead to neuronal death via upregulated ROS species and oxidative stress.36–37 During the hypoxic condition, ROS species can be produced at higher amounts during electron transport chain process or uncoupled nitric oxide synthase process; the upregulated form of ROS species could result in cellular damage, as a defence mechanism the oxidative stress activates the antioxidant genes such as FOXO or SOD2 to counter the overexpressed ROS species. The imbalance between the antioxidant genes and ROS level can result in lipid oxidation, DNA degradation and tissue damage causing cellular dysfunction; hypoxic stress disrupts the redox signalling in the cells that cause dysfunctioning of various cellular pathways, molecular damage and apoptosis.38–39 Under hypoxic stress, the ROS level could be downregulated via targeting mitochondrial electron transport chain, inhibiting the complex III that may help in the downregulation of ROS level; enhancing antioxidant defence mechanism such as SOD2 and FOXO can help in maintaining redox balance. Similarly, the modulation of HIFs can inhibit the synthase of ROS levels. By working into these highlighted issues between hypoxia, mitochondrial dysfunction and alpha-synuclein pathology, we can develop better therapeutic confirmations and offer more effective neuroprotective effects.
Strengths of the Studies
The studies highlight the relationship between alpha-synuclein and mitochondrial dysfunction in PD and the role of hypoxia as an intermediate factor. The interaction between alpha-synuclein and mitochondria, particularly through membranal lipids like CL, is emphasized as a key factor in mitochondrial disruption and neurodegeneration. The studies discuss the involvement of other proteins, such as peroxisome proliferator-activated receptor gamma coactivator, Sirtuin-1, Sirtuin-3 and adenosine monophosphate (AMP)-activated protein kinase, in maintaining mitochondrial biogenesis during hypoxia. Hypoxia is identified as a crucial link between alpha-synuclein and mitochondrial regulation, leading to neuronal death in PD.
Weaknesses and Areas of Controversy or Uncertainty
The primary player in inducing the other, alpha-synuclein or mitochondria, is still debatable. The cause and interlinked mechanisms of PD, including the role of hypoxia, alpha-synuclein and mitochondrial dysfunction, remain unclear. Limited clinical approaches and the lack of specific point mutations in PD models make it challenging to identify the appropriate molecular mechanisms for halting mitochondrial degradation. The complexity of pathophysiology and the potential disturbance or initiation of other cellular pathways by modulating the factors involved make it difficult to translate insights into clinical practice. Developing specific biomarkers like alpha-synuclein for early diagnosis and effective treatments in PD is a challenging task.
Conclusion
The present scenario shows expansive thriving treatment towards PD, but ongoing clinical trials have still many unclear doubts such as the pathologies involved in the progression of PD. Limited clinical approaches have shown some potential mechanisms, but the cause and interlinked mechanisms remain unclear. Among these, we gather that hypoxic-based oxidative stress could be the one possible mechanism inducing alpha-synuclein in disrupting mitochondrial biogenesis. We have discussed how alpha-synuclein aggregation is achieved via oxidative stress and producing ROS, which leads to neurodegeneration. The current study on the subject also notes that there are no particular point mutations in PD models to identify the appropriate molecular mechanisms in halting further mitochondrial degradation. Overexpressed alpha-synuclein provides toxicity to neurons via mitochondrial dysfunction. Hypoxia makes the brain more susceptible to brain injury even death in the case of cerebral hypoxia. Hypoxia is a signal to the brain that there is not enough oxygen in the blood, which leads to many lipid and protein rearrangements and other neuronal defects. The brain cannot withstand hypoxia for an extended period; in this instance, it may result in oxidative stress, apoptosis and an electron transport chain breakdown that lowers ATP production. The more rapid development of mitochondrial dysfunction and neuronal death is caused by the overproduction of ROS, such as free hydroxyls and peroxides, but it is still unclear how hypoxia can be utilized as a crucial marker to identify further mitochondrial degradation. HIFs are mainly involved when there is a low supply of oxygen as it is involved in other activities such as vascularization and erythropoietin findings suggest that activation of HIFs could help in cell survival during hypoxic stress. The brain is more susceptible to oxygen supply because of its increased energy demand, which can lead to hypoxic injury. The hypoxic injury could be also regulated by activating HIFs to gain stability. HIFs are also involved in mitochondrial biogenesis, by increasing the levels of HIFs by inhibiting the prolyl hydroxylase (a mechanism that detects low pressure of oxygen or hypoxic conditions within the cells) can result in a neuroprotective effect. 40 We can also focus on other proteins such as peroxisome proliferator-activated receptor gamma coactivator, Sirtuin-1, Sirtuin-3 and AMP-activated protein kinase that are involved in mitochondria during hypoxia which upon regulation might show an efficient role in maintaining the mitochondrial biogenesis; the activation or overexpression of sirtuin1/3 help in the coactivation of peroxisome proliferator-activated receptor gamma coactivator, glutathione peroxidase and antioxidants genes (FOXO and SOD2), where all these genes are involved in the downregulation of ROS species and oxidative stress which result in mitochondrial biogenesis or neuroprotection.41–43. Translating these insights into the clinical practice between hypoxia, mitochondria and alpha-synuclein may be a back-breaking/challenging task due to the complexity of pathophysiology where the modulation of the factors could disturb or initiate other cellular pathways. Developing specific biomarkers like alpha-synuclein could be a challenging task in clinical practice. The insights also bring breaking opportunities by early diagnosis of alpha-synuclein as a potential biomarker offering patient stratification, and monitoring disease progression in PD. Deeper studies on hypoxia, alpha-synuclein and mitochondria could yield more effective treatments for PD. On this note, this study will emphasize the gaps and highlight the hypothetical approaches that are leading to severe neurodegeneration via hypoxic injuries, and we will be able to present some potential therapies.
Abbreviations
Footnotes
Acknowledgments
We acknowledge the generous research infrastructure and support from JSS College of Pharmacy, JSS Academy of Higher Education & Research, Rocklands, Ooty, The Nilgiris, Tamil Nadu, India.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical Approval
Not applicable.
Informed Consent
Not applicable.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.