
Abstract
Alpha-dicarbonyls such as glyoxal (GO) trigger mitochondrial dysfunction resulting in the development of different diabetic complications. The present study investigated the effects of lovastatin against GO-induced toxicity on rat liver mitochondria. The rat liver mitochondria (0.5 mg protein/mL) were treated with various concentrations of lovastatin (1, 5, 10 µM) at 37°C for 30 min and then exposed to GO (3 mM) at 37°C for 30 min. Oxidative stress markers including MDA, reactive oxygen species (ROS), glutathione (GSH) and protein carbonylation (PC) level were measured. Mitochondrial complex II activity and mitochondrial membrane potential (MMP) were assessed for evaluating mitochondrial function. Glyoxal significantly increased the level of ROS, PC and MDA. This effect was associated with the reduction of MMP, complex II activity and GSH content. Pre-treatment with lovastatin potentially reversed GO-induced mitochondrial toxicity. These results suggest that lovastatin have a protective effect against GO-induced toxicity in isolated rat liver mitochondria.
Introduction
Diabetes mellitus (DM) is a chronic disease, which results from the abnormality in the metabolism of glucose leading to the glucose intolerance and hyperglycemia. This metabolic disorder occurs when the pancreas is unable to generate sufficient amounts of insulin or when the cells of target tissues fail to respond to insulin. 1 Hyperglycemia induces oxidative stress and activates various signaling pathways leading to the development of different diabetic complications including micro- (nephropathy, neuropathy and retinopathy) and macro-vascular (ischemic heart disease, peripheral vascular disease, and cerebrovascular disease) complications2,3; these complications are the main causes of mortality and morbidity in diabetic patients with poor blood glucose control. 4 In tissues, glucose is autoxidized to form α-dicarbonyl compounds such as glyoxal (GO) and methylglyoxal (MG). These reactive dicarbonyls cause non-enzymatic glycation of cellular compounds such as proteins, DNA and lipids resulting in the formation of advanced glycation end products (AGEs); these compounds cause a number of cellular damages and they are linked to the development of diabetic complications.5,6 Glyoxal has destructive effects on mitochondrial function which this is associated with the impairment of mitochondrial electron transport chain function; this effect leads to the elevation of reactive oxygen species (ROS) generation, reduction of antioxidant level and disruption of mitochondrial membrane potential (MMP). 7 Excessive generation of ROS is associated with the induction of NFκB/TNF-α signaling pathway leading to the activation of inflammatory responses in various tissues such as liver.8–10 Therefore, agents with antioxidant properties can reduce GO-mediated generation of ROS in the mitochondria, resulting in the prevention of oxidative stress-induced cellular damage. 4
Lovastatin is a natural drug, which belongs to statins 11 ; this group of drugs acts by competitive inhibition of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase. HMG-CoA reductase is a rate-limiting enzyme in the cholesterol biosynthetic pathway, catalyzing the conversion of HMG-CoA to mevalonate. Although the primary reason for using statins is the reduction of serum cholesterol levels, statins have noncholesterol-lowering or pleotropic effects including antioxidant, anti-inflammatory and immunomodulatory properties. 12 Lovastatin is reported to reduce ROS-mediated stress through down-regulating NADPH oxidase expression. 13 Furthermore, lovastatin increases the activity of antioxidant enzymes including catalase and glutathione peroxidase and level of reduced glutathione (GSH) in various tissues. 14 Lovastatin stabilizes the mitochondrial membrane in oxidative stress condition resulting in the maintenance of ATP production by mitochondria. 15 In experimental autoimmune encephalomyelitis (EAE), lovastatin in combination with 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR) attenuates the EAE disease through protecting against loss of mitochondria (complex I and citrate synthase) as well as levels of ATP. 16 Considering the important etiological role of reactive dicarbonyls such as GO in the induction of oxidative stress and mitochondrial dysfunction and on the other hand the antioxidant properties of lovastatin, this hypothesis has been raised that lovastatin may inhibit GO-induced oxidative stress. Current work was, therefore, carried out to investigate the ability of lovastatin to prevent GO-induced oxidative stress in isolated rat liver mitochondria.
Material and methods
Chemicals
Glyoxal (GO, 40% w/v), lovastatin and other reagents were obtained from Sigma- Aldrich Chemical Company (St. Louis, MO, U.S.A).
Animals
Adult male Wistar rats (200 ± 20 g) were obtained from the animal house of Ahvaz Jundishapur University of Medical Sciences. Animals had free access to water and standard chow diet and were maintained under standard laboratory condition (a controlled temperature (25 ± 2°C) with a 12-h light/dark cycle). All experimental procedures were performed in accordance with the Animal Ethics Committee Guidelines (ethics approval number: IR.AJUMS.REC.1396.666).
Mitochondrial extraction
Mitochondria were extracted from the liver tissue of Wistar rats. 17 Animals were deeply anesthetized with a mixture of ketamine/xylazine (100/10 mg/kg, i.p.), and then sacrificed by decapitation and liver tissues were removed immediately. Isolated liver tissues were minced in a fresh and cold mannitol solution (pH 7.4) containing d-mannitol (200 mM), sucrose (70 mM), EGTA (1 mM), BSA (0.1% (w/v)), HEPES-KOH (10 mM). The tissue solution was homogenized and then centrifuged at 600×g for 10 min at 4°C for the sedimention of nuclei and broken cell debris. The supernatant was collected and centrifuged at 10,000×g for 15 min. Pellet was re-suspended in the isolation medium and was then centrifuged at 10,000×g for 10 min. The achieved mitochondrial pellet was re-suspended in the mannitol buffer and the protein concentration was determined using Bradford assay, 1976. 18
Experimental design
The suspensions of the mitochondria were treated with the various concentrations of lovastatin (1, 5 and 10 µM) for 30 min and then incubated with GO (3 mM) at 37°C for 30 min.
Measurement of complex II activity (viability)
The activity of mitochondrial complex II was assessed using the measurement of succinate dehydrogenase enzyme activity by MTT assay. 19 The mitochondrial solution (0.5 mg protein/ml) was centrifuged (at 10,000 ×g for 1 min) and the achieved pellet was then re-suspended in isolation medium and MTT solution. After incubation of mixture at 37°C for 45 min, produced purple formazan crystals were dissolved in DMSO and the optical density was measured at 570 nm using Synergy HT Microplate Reader (BioTek Instruments, Inc, Winooski, VT, USA).
Lipid peroxidation assay
For the determination of lipid peroxidation level in the mitochondrial solution, the amount of malondialdehyde (MDA) was measured using the MDA assay kit (Teb Pazhouhan Razi (TPR), Tehran, IRAN). Butylated hydroxytoluene was added to mitochondrial suspensions (0.5 mg protein/ml) for the prevention of further peroxidation. Mitochondrial suspensions were then mixed with detergent and chromogenic solution (thiobarbituric acid, alkali, and acetic acid). Tubes were boiled in water for 60 min to appear a pink color. Tubes were cooled at ice-bath and centrifuged at 3500 ×g for 10 min. The supernatants of samples were collected and the absorbance was measured at 532 nm using Synergy HT Microplate Reader (BioTek Instruments, Inc, Winooski, VT, USA). The MDA standard curve was used to determine of MDA level in samples. The content of MDA was expressed as nmol/mg protein.
Mitochondrial ROS level
The fluorescent probe DCF-DA was used to measure the level of mitochondrial ROS. 20 The solution of DCF-DA (1.6 µM) was added to mitochondrial suspensions (0.5 mg protein/ml) and incubated at 37°C for 10 min. The fluorescence was measured using 500 nm excitation and 520 nm emission wavelength by Perkin Elmer LS-50B Luminescence fluorescence spectrophotometer (California, United Stated).
Measurement of MMP
Mitochondrial membrane potential was evaluated by measuring the amount of cationic fluorescent dye, Rh 123, uptake by mitochondria. 21 Briefly, mitochondrial suspensions (0.5 mg protein/ml) were mixed with Rh 123 (1.5 µM) and shaken at 37°C for 10 min. The fluorescence was measured using 490 nm excitation and 535 nm emission wavelength by Perkin Elmer LS-50B Luminescence fluorescence spectrophotometer (CA, USA).
Measurement of GSH content
The mitochondrial GSH content was determined as previously described. 22 The mitochondrial suspensions (0.5 mg protein/ml) were mixed with Ellman’s reagent (DTNB; 0.04% in 0.1 mol/L of phosphate buffers (pH 7.4)). The obtained mixture was incubated at room temperature for 20 min to obtain the yellow color. The optical density of developed color was read at 412 nm using a microplate reader (BioTek Instruments, Inc, Winooski, VT, USA). The GSH standard curve was used to determine of GSH level in samples. The content of GSH was expressed as µg/mg protein.
Protein carbonylation assay
The protein oxidation in mitochondrial suspensions was determined by measuring the total protein-bound carbonyl (PC) level. 23 To determine the PC level in mitochondrial suspensions, a mixture of mitochondrial suspensions (0.5 mg protein/ml, 0.5 mL) and 0.1% DNPH (w/v in 2 N HCl; 0.5 mL) was incubated at room temperature for 1 h. Ice-cold 20% TCA (v/v; 1 mL) was added to mixture and centrifuged at 10,000 g for 10 min to precipitate total protein. For extraction of free DNPH, the obtained pellet was washed thrice with an excess volume of ethanol: ethyl acetate (1:1) solution. The extracted mitochondrial protein was dried under a stream of nitrogen and then dissolved in tris buffer solution containing 8.0 M guanidine hydrochloride (pH 7.2). The absorbance of solubilized hydrazones was measured using a microplate reader (BioTek Instruments, Inc, Winooski, VT, USA) at 375 nm and expressed as nmol/mg protein.
Statistical analyses
Statistical analysis was performed using Graph Pad Prism version 6 for Windows (Graph Pad Software, USA). Results were presented as mean ± standard deviations (SD). Individual differences between the groups were determined using one-way ANOVA followed by Tukey’s post hoc test and p < 0.05 was considered statistically significant.
Results
The effect of lovastatin on GO-induced alteration in MMP and the activity of mitochondrial complex II
Results showed that GO (3 mM) significantly reduced the activity of succinate dehydrogenase in the mitochondria (p < 0.05). Treatment of mitochondrial suspensions with lovastatin (5 and 10 µM) caused a significant increase in the mitochondrial viability compared to GO-treated group (p < 0.05, Figure 1(A)).
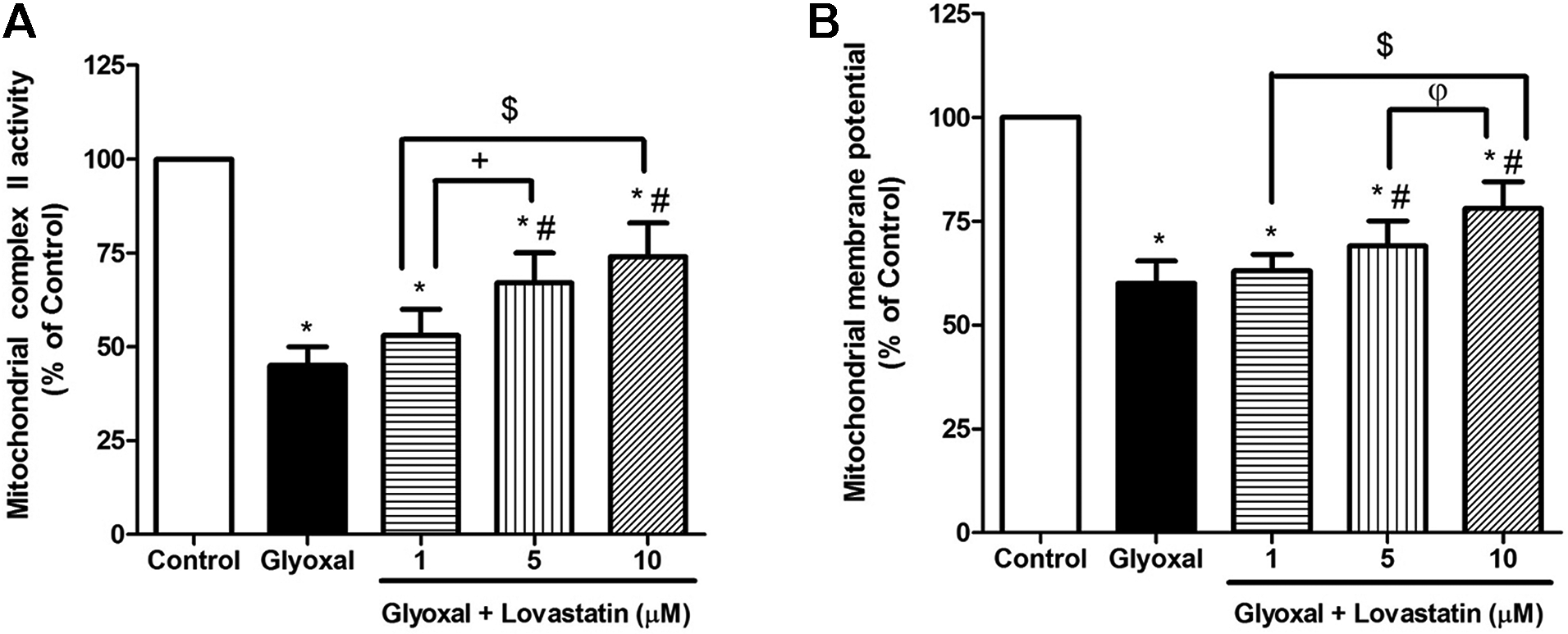
(A) The effect of lovastatin on GO-induced alteration in mitochondrial complex II activity. (B) The effect of lovastatin on GO-induced alteration MMP. Values were expressed as mean ± SD (n = 7). *p < 0.05 as compared to the control group. #p < 0.05 as compared to the GO-treated group.
The result from Rh123 staining showed that the exposure of mitochondrial suspensions with GO (3 mM) significantly reduced MMP (p < 0.05). Treatment with lovastatin (5 and 10 µM) significantly inhibited MMP collapse compared to GO-treated group (p < 0.05, Figure 1(B)).
The effect of lovastatin on oxidative stress in GO-induced mitochondria
The level of ROS, MDA and PC significantly increased in GO (3 mM)-treated group compared to control group (p < 0.05). Treatment with lovastatin (1, 5 and 10 µM) significantly reduced the GO-induced elevated level of MDA and PC (p < 0.05, Figures 2(B) and 3(A)). Lovastatin in 10 µM also significantly decreased GO-induced elevation level of ROS (p < 0.05, Figure 2(A)). Current results indicated that the exposure of mitochondria with GO (3 mM) significantly reduced the level of GSH compared to the control group (p < 0.05). Treatment with lovastatin (5 and 10 µM) significantly inhibited GO-induced mitochondrial GSH oxidation (p < 0.05, Figure 3(B)).

(A and B) The effect of lovastatin on GO-induced alteration in the level of mitochondrial ROS and MDA levels. Values were represented as mean ± SD (n = 7). *p < 0.05 as compared to the control group. #p < 0.05 as compared to the GO-treated group.

(A and B) The effect of lovastatin on GO-induced alteration in the level of mitochondrial PC and GSH levels. Values were represented as mean ± SD (n = 7). *p < 0.05 as compared to the control group. #p < 0.05 as compared to the GO-treated group.
Discussion
The present study was investigated the effects of lovastatin (1, 5 and 10 µM) against GO (3 mM)-induced mitochondrial oxidative stress and alteration in mitochondrial function in the mitochondria extracted from rat liver. Hyperglycemia in diabetic patients is associated with the over-production of ROS leading to the autoxidation of glucose to α-dicarbonyl compounds such as GO; these reactive dicarbonyls cause auto-oxidation of glucose, lipids and amino acids of proteins resulting in the formation of carbonyl functional groups. 24 Reactive carbonyl compounds also trigger mitochondrial dysfunction resulting in the excessive generation of ROS, the loss of mitochondrial antioxidant capacity and disruption of MMP. 7 Furthermore, ROS directly react with various cellular components including proteins, lipids, DNA, membranes of cells and cellular organelles leading to the oxidation of lipids and proteins. Lipid peroxidation results in the formation of various secondary products such as aldehydes; among these aldehydes, MDA is recognized as the most popular biomarker for oxidative damage. 25 In line with previous studies, 7 our results showed that exposure of rat liver mitochondria with GO increased the generation of ROS, MDA and carbonyl proteins, which this effect was associated with the depletion of mitochondrial antioxidant capacity determined by the reduction of GSH level. Our in vitro experiments also revealed that treatment of mitochondrial suspensions with lovastatin prior to incubation with GO reduced the generation of ROS, MDA and PC and increased the GSH content. These findings are in agreement with previous studies indicating the antioxidant effect of lovastatin. Lovastatin has been reported to reduce oxidative stress in rats with multiple cardiovascular risk factors. This effect of lovastatin is mediated by the up-regulation of miR-29b expression through inducing the AMPK phosphorylation in endothelial cells. Over-expression of miR-29b leads to the lovastatin-induced suppression of PA200, a proteasome activator, resulting in the inhibition of proteasome activity. Activation of proteasome induces oxidative stress in endothelial cells through degrading IKBα or GTP cyclohydrolase I, therefore, suppression of proteasome activity by lovastatin results in the inhibition of oxidative stress in endothelial cells. 26 Lovastatin increases the level of GSH and the activity of antioxidant enzymes including glutathione-S-transferase (GST), superoxide dismutase (SOD) in the liver of diabetic rats. 27 The stimulatory effect of lovastatin on the antioxidant enzymes activities may result from its ability to induce the activation of nuclear translocation of NF-E2-related factor-2 (Nrf2) transcription factor, a transcription factor regulating the expression of antioxidant proteins. 28
The current study indicated that exposure of mitochondria with GO resulted in the impairment of mitochondrial function characterized by the disruption of succinate dehydrogenase (complex II) activity and MMP. Mitochondria are the main source of ROS production in the cell. 29 The activity of complexes I (NADH-ubiquinone oxidoreductase) and III (ubiquinol-cytochrome c oxidoreductase) results in the generation of ROS 30 ; however, the role of complex II in the generation of ROS is negligible. Co-enzyme Q10 transfers electron from complex I and complex II to complex III; reduced co-enzyme Q10 is a potent antioxidant, which inhibits mitochondrial superoxide production and protects mitochondrial lipids and proteins from oxidative stress-induced damage. 31 Therefore, the impairment of complex II results in the elevation of oxidized co-enzyme Q10 level and subsequent accumulation of ROS in the mitochondria. 30 Cardiolipin is one of mitochondrial lipids located in the inner mitochondrial membrane; because of the presence of high content of unsaturated fatty acids in cardiolipin and its location in the near to the site of ROS production, it is suggested that cardiolipin be a primary target of mitochondrial ROS. Destructive effects of ROS on the mitochondrial membrane lipids and proteins leads to the mitochondrial membrane disruption and subsequent loss of MMP. 32 The reduction of MMP is a hallmark of mitochondrial dysfunction, triggering a series of events including cytochrome c release and activation of caspases resulting in the induction of apoptosis. 33 This investigation demonstrated that lovastatin inhibited GO-induced destructive effect on mitochondrial function through improving the activity of complex II and MMP. In agreement with these findings, previous studies have reported the protective effect of lovastatin on mitochondria against various pathologic conditions. In the ischemia-reperfusion condition in kidneys of rats, lovastatin has been reported to protect mitochondrial function leading to the maintenance of ATP production. 15 Lovastatin prevents from the loss of MMP in mesenchymal stem cells under conditions of hypoxia and serum deprivation leading to the inhibition of mitochondrial apoptosis; this effect results from the activation of PI3K/Akt and MEK/ERK1/2signaling pathways. 34
Conclusions
Glyoxal (GO) caused mitochondrial toxicity characterized by the impairment of succinate dehydrogenase enzyme activity, the loss of MMP, and the elevation of ROS, MDA and PC production. These effects were associated with the reduction of GSH content. Our results indicated that lovastatin is able to inhibit GO-induced mitochondrial dysfunction; this effect may result from the reduction of oxidative stress in rat liver mitochondria. These results suggest that lovastatin may be an agent to prevent GO-induced mitochondrial toxicity.
Footnotes
Author contributions
Conception and design: M.G.: performing the literature search, data collection and analysis: A.H., M.R., M.B., A.N. and S.M.: drafting the manuscript: all authors. Approving the final version: all authors. M.G. is responsible for the integrity of the work as a whole.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval and consent to participate
The experimental procedures were performed according to the Animal Ethics Committee Guidelines for the use of experimental animals in research (Ethic code: IR.AJUMS.REC.1396.666).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Deputy of Research of Jundishapur University of Medical Sciences, Ahvaz, Iran under grant number APRC-9610.