
Abstract
Objective:
To computationally model the CTX-M-5 β-lactamase and establish its structure, which is exclusively present in human-associated Salmonella.
Methods:
The CTX-M-5 aminoacid sequence (Uniprot ID:O65975) of Salmonella enterica subsp. enterica serovar typhimurium was retrieved from UniProt database and subjected to homology modeling using MODELLER 9v7. The homology models were duly validated using RAMPAGE tool by generating Ramachandran plots, ERRAT graphs, and ProSA score. DoGSiteScorer server and ConSurf server were used to detect the cavities, pockets, and clefts to identify conserved amino acid sites in the predicted model. Subsequently, the modeled structure was docked using CLC Drug Discovery Workbench against proven drugs and known inhibitors.
Results:
Obtained high-quality homology model with 91.7% of the residues in favorable regions in Ramachandran plot and qualified in other quality parameters. Docking studies resulted in a higher dock score for PNK (D-benzylpenicilloic acid) molecule when compared to other reported inhibitors.
Conclusion:
This in silico study suggests that the compound PNK could be an efficient ligand for CTX-M-5 β-lactamase and serve as a potent inhibitor of CTX-M-5.
Introduction
Salmonella enterica subsp. enterica serovar typhimurium (aka S. typhimurium), a member of the Enterobacteriaceae family, is a gram-negative, facultative anaerobe that causes self-limiting gastroenteritis in humans. On the other hand, infection is systemic in murine models that resemble typhoid fever in humans caused by Salmonella enterica subsp. enterica serovar typhi. Salmonella is one of the four major causes of diarrheal diseases and foodborne infections across the globe.
1
The World Health Organization reports 93,000,000 cases of enteric infections and 155,000 mortality cases annually.
2
Currently, drugs used in the treatment of gastroenteritis lead to the increasing emergence of antibiotic resistance and the most rampant antibiotic-resistant mechanism is the production of β-lactamases by gram-negative bacteria.
3
β-lactamases inactivate β-lactam group of antibiotics by hydrolyzing the amide bond of a four-membered β-lactam ring. Based on sequence variation, about 4300 different types of β-lactamases have been identified so far (
Extended-spectrum β-lactamases (ESBLs) are plasmid-mediated β-lactamases that provide resistance to the extended-spectrum group of cephalosporins and other closely related oxyiminocephalosporins namely ceftazidime, ceftriaxone, cefotaxime, and monobactam antibiotics such as aztreonam. However, they are susceptible to carbapenem antibiotics (imipenem, meropenem, and ertapenem), cephamycins, and β-lactamase inhibitors, such as clavulanic acid and ethylenediaminetetraacetic acid. Ambler’s classification illustrated ESBL enzymes such as SHV, TEM, CTX-M, VEB, and GES as class A β-lactamases and OXA-ESBLs as class D β-lactamases.
5
Point mutations in the active site of known β-lactamases such as TEM, SHV, and OXA-10 led to the evolution of new β-lactamases called ESBLs. There were 193 SHV types and 223 TEM types of ESBLs as detailed in Beta-Lactamase Database (
Although TEM and SHV ESBLs were more predominant during 1980s, in the recent past, CTX-Ms had completely supplanted them. At present, CTX-Ms have completely strewn the world referring as “CTX-M pandemic.”7, 8 Being isolated from Munich and highly active on cefotaxime, they are named as CTX-Ms. 9 Currently, there are about 172 different varieties of CTX-M-β-lactamases reported in the literature. According to Bush and Ambler’s class A classification, CTX-Ms were clustered into five individual groups, such as CTX-M-1, CTX-M-2, CTX-M-8, CTX-M-9, and CTX-M-25, named after the first member of each group. 6 Clustering of CTX-M-5 in CTX-M-2 group was first reported in S. typhimurium. 10 Later a widespread occurrence of CTX-M-5 across the world was observed.11–15 CTX-M-5 β-lactamase hydrolyses cefotaxime more actively than ceftazidimes, as methoxy iminogroup of cefotaxime provide better affinity to the enzyme than carboxy-isopropoximino group of ceftazidimes. 9
CTX-M-2, CTX-M-3, CTX-M-9, CTX-M-14, and CTX-M-15 are found in both animal and human Salmonella isolates, whereas CTX-M-5 ESBLs are found only in human Salmonella isolates.10, 16, 17 Members of CTX-M group such as CTXM-9, 18 CTX-M-15, CTX-M-53, CTX-M-71, CTX-M-82, and CTX-M-89, 19 were docked with different drugs and inhibitors. However, the information about three-dimensional structure and functional analysis of the CTX-M-5 was not available so far. As CTX-M-5 is rapidly evolving in Salmonella sps. across the world, there is an utmost need to model CTX-M-5 in silico and target its active site to combat its widespread. A 3D model will help to determine the active sites of protein for docking studies. Thus, modeling a structure using a computational biology approach is of enormous advantage as it consumes less time and is cost effective. Hence, the present study was carried out to predict a 3D structure for CTX-M-5β-lactamases protein, which is specific to human Salmonella isolates. Additionally, we focused on the evaluation of 3D structure and identification of conserved residue sites and ligand binding sites. Thus, the objectives of the present study are (a) homology modeling of CTX-M-5, (b) phylogenetic analysis of CTX-M-5, (c) evaluation of 3D structure, (d) assessment of functional sites in CTX-M-5, and (e) docking of CTX-M-5 with different drugs and inhibitors.
Materials and Methods
Data Retrieval and Template Selection
CTX-M-5 β-lactamase amino acid sequence of Salmonella enterica subsps. enterica serovar typhimurium (Uniprot ID: O65975) was retrieved from UniProt database. To find out suitable templates for homology modeling, blaCTX-M-5 was subjected to BLAST-P search against Protein Data Bank (PDB).
Phylogenetic Analysis
Phylogenetic analysis was carried out for a total of eight amino acid sequences using MEGA version 7. 20 Neighbor-joining method was employed to know the evolutionary history. The evolutionary distances were computed through the Poisson correction method. The bootstrap value of 1000 was used for the analysis of evolutionary history. Gaps and missing data were eliminated from positions.
Homology Modeling and Validation
The three dimensional structures for CTX-M-5 were predicted using MODELLER 9v7. The predicted structures were evaluated using RAMPAGE (Ramachandran plot), 21 ERRAT graphs (Colovos and Yeates 1993) 22 , and ProSA analysis. 23
Functional Site Analysis
DoGSiteScorer server 24 was used to predict the cavities, pockets, and clefts. ConSurf server was used to determine the functional and conserved residue regions of the desired protein sequence with respect to other homologous sequences.25, 26 COACH, a meta server, was employed for determining ligand binding sites in the protein. 27
Molecular Docking
The predicted best structure of CTX-M-5 was docked with different drugs and inhibitors. The ligand molecules were retrieved from ZINC database (
Results
Data Retrieval and Template Selection
A complete amino acid sequence (Uniprot ID: O65975) of CTX-M-5 β-lactamase (EC: 3.5.2.6) of Salmonella enterica subsps. enterica serovar typhimurium with 291 amino acid residues length was retrieved from the UniProt database (O65975). CTX-M-5 β-lactamase protein sequence was subjected to PDB BLAST to identify structural homologs. The hit list consisted of chain A, β-lactamase Toho-1 of Escherichia coli (PDB: 4X69_A; structure resolution: 1.42, identity: 97.32%, coverage: 89%), chain A, β-lactamase Toho-1 of E. coli (PDB: 1IYS_A; structure resolution: 1.65, identity: 97.32%, coverage: 89%), chain A, β-lactamase Toho-1 of E. coli BL21 (PDB: 4BD0_A, structure resolution: 1.2, identity: 96.93%, coverage: 89%), and chain A, apo structure of class A β-lactamase Toho-1 of E. coli (PDB: 2ZQ8_A, structure resolution: 1.03, identity: 96.95%, coverage: 90 %). Amongst them, protein bearing PDBID: 2ZQ8_A was selected as a template for homology modeling of the target protein as it has a better structural resolution (1.03) compared to other proteins. Furthermore, the evolutionary analysis showed that 2ZQ8_A, i.e., Toho-1 of E. coli, and CTX-M-5 of S. typhimurium clustered together in the phylogenetic tree, demonstrating their evolutionary relatedness and justifying the selection of 2ZQ8_A as a template for the prediction of CTX-M-5 model. Phylogenetic analysis (Figure 1) revealed the evolutionary relationship of CTX-Ms and reported the average rate of divergence as 0.02.
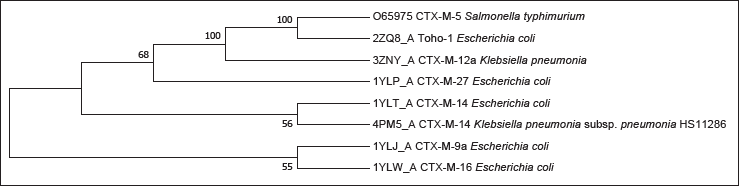
Phylogenetic analysis was performed in MEGA7, using the neighbor-joining method. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. 2
Homology Modeling and Validation
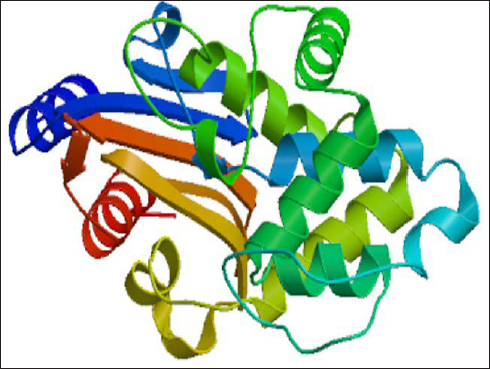
Homology modeling intends to build a 3D structure of a protein whose structure is experimentally unknown. It utilizes the structures of experimentally proven and verified structures of relevant protein family members as templates for prediction. MODELLER 9v7 was used to predict the 3D structure (Figure 2) for CTX-M-5 by using 2ZQ8_A (PDB ID) as the template and the resulting structure was visualized using RasMol (Bernstein 2000). 28 A total of 100 3D structures were generated and the structure with the lowest discrete optimized protein energy score was chosen for further analysis. The molecule, (No: 62nd) with best DOPE score –30663.2 and GA341 score, 1.0 was selected for further studies. The total energy before and after energy minimization was –6143.1 kJ/mol and –13845 kJ/mol, respectively, and energy minimization was accomplished with GROMOS96 43B1 using Swiss-Pdb Viewer.
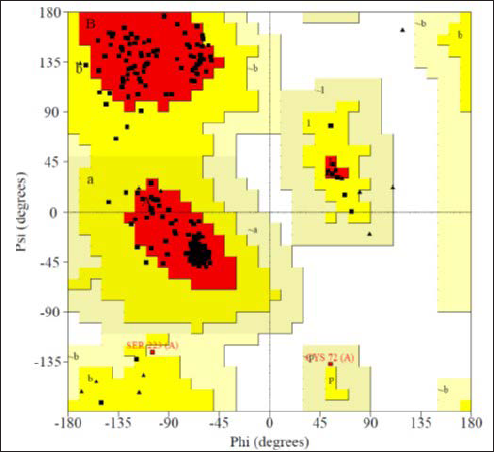
RAMPAGE analysis for the modeled CTXM-5 protein revealed that 91.7% of the residues in the allowed regions confirming the accuracy of the modeled structure (Figure 3). The Ramachandran plot displayed the statistical distribution of the combinations of the backbone dihedral angles ϕ and ψ that are favorable, unfavorable, and disallowed. The permitted regions of the plot represent the values of ϕ/ψ angles that are possible for an amino acid. Structure validation is based on the dispersion of ϕ/ψ values in a protein structure. In the Ramachandran plot, dihedral angles observed in the forbidden regions denote low-quality homology models representing the structural problem. In the current model, all the favored and allowed regions resemble the conformations of the amino acids and reported no steric hindrance. A good quality model would be expected to have a threshold value of over 90% in the most favored regions.
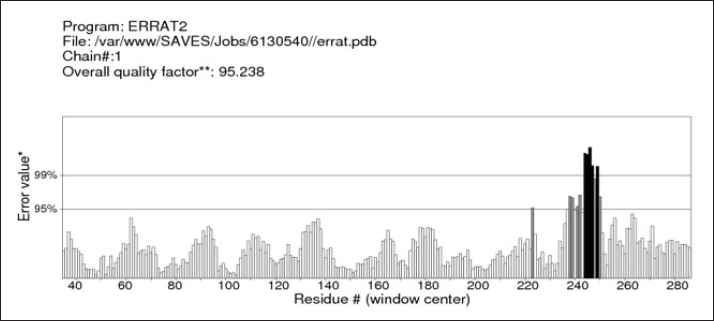
Verification of protein structure by ERRAT analysis exhibited an overall quality factor of 95.238 (Figure 4), which depicts a good 3D structure of the predicted model. In addition to this, it also examines the statistics of nonbonded atomic interactions. It is the graph of the error value function plotted against the position of a nine-residue sliding window, calculated by comparing data from highly refined structures with each other. Unrefined structures generally do not score well and the accepted range is >50 for a high-quality model.
The ProSA Z-score indicates the overall model quality of the predicted 3D structure. Previous studies suggest that the Z-score of query protein should be within the range of our template protein inorder to be considered a good quality score. The Z-score of the predicted protein (–7.63) is within the range of our template protein (–7.88) and hence the modeled structure is a valid one (Figure 5).
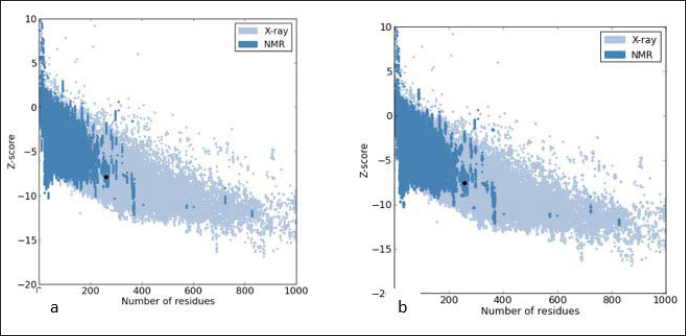
ProSA-web Z-scores of all protein chains in PDB are are denoted by dots as determined by X-ray crystallography (blue dots) with respect to their length (number of residues). (a) The Z-score of proteins is highlighted (black dot). The Z-score of protein 2zq8 is –7.88. (b) The Z-score of the predicted structure is –7.63.
Functional Site Analysis
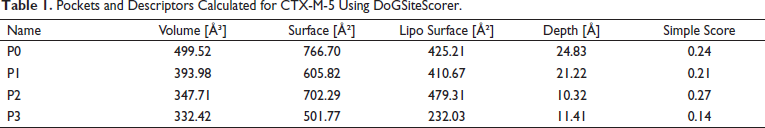
DoGSiteScorer determines the potential active sites of a 3D protein structure of CTX-M-5. It uses a difference of Gaussian filter to detect potential pockets on the protein surface and splits them into subpockets, describing their size, shape, and chemical features in the given query protein structure. The higher the score, the more likely the pocket is to be druggable. Pocket P2 reported the higher score (Table 1) with a depth of about 10.32 Å and the volume of about 347.71 Å 3 . It is predicted that pocket P2 contains the highest lipophilic surface when compared to other pockets, i.e., about 479.31 Å 2 comprising alanine and leucine as predominant amino acids in its pocket. Hence, P2 appears to be a potential active site for CTX-M-5.
Pockets and Descriptors Calculated for CTX-M-5 Using DoGSiteScorer.
Conserved Sites Determination
The ConSurf server was used for identification of evolutionarily conserved amino acid residues in our predicted protein molecule. It uses the empirical Bayesian method for the calculation of conservation scores. 29 This method gives accurate conservation scores when compared to others’ even when very small sequences are used. The conserved sites of CTX-M-5 were displayed both in sequence as well as in the 3D structure (Figure 6). Results were interpreted on a conservation scale of levels ranging from 1 to 9 represented with different colors. Levels 1 and 2 (blue) depict the variable region, level 5 (white) show average score, and levels 8 and 9 (bordeaux) show moderately conserved and highly conserved regions. According to the above analysis, CTX-M-5 protein has 65 highly conserved functional residues exposed and 14 highly conserved structural residues buried that contribute to the active sites of the protein. Thus, we suggest that this method of identification of conserved functional residues is merely helpful with respect to other methods in understanding the 3D structure of the protein.

Protein–Ligand Binding Site Prediction
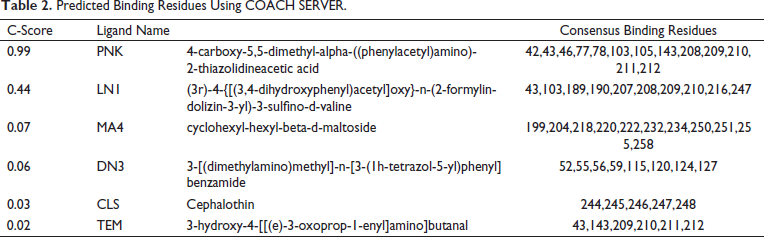
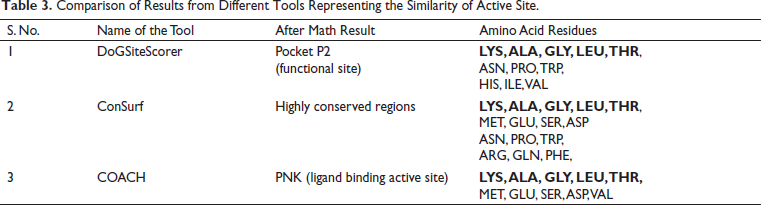
COACH server was used to determine the ligand-binding sites besides predicting an efficient ligand that can bind CTX-M-5 (Table 2). It generates complementary sites by searching against BioLip protein function database. C-score is the confidential score of the COACH server ranging from 0 to 1, where a PNK molecule with a 0.99 score stands as a reliable compound. The binding residues of CTX-M-5 from different bioinformatics tools like DoGSiteScorer, ConSurf, and COACH servers were compared (Table 3). From the analysis, Lys, Ala, Gly, Leu, and Thr were found to be common residues represented in bold font in Table 3 that could form hydrogen bond interactions with the drugs and inhibitors.
Predicted Binding Residues Using COACH SERVER.
Comparison of Results from Different Tools Representing the Similarity of Active Site.
Docking Analysis
Modeled CTX-M-5 was docked with different inhibitors (clavulanic acid, sulbactam, and tazobactam) and drugs (cefotaxime, cefipime, and cefixime) along with PNK molecule using CLC Drug discovery Workbench (Figure 7). The binding sites include a center binding pocket of 138.75 Å 3 and the number of iterations was set to 100. For each ligand, five docking outcomes were returned. When the protein and ligand interact, the CLC score simulates the possible energy change. A high negative value indicates a strong binding, whereas a lower negative or positive score indicates a weak binding (Table 4).
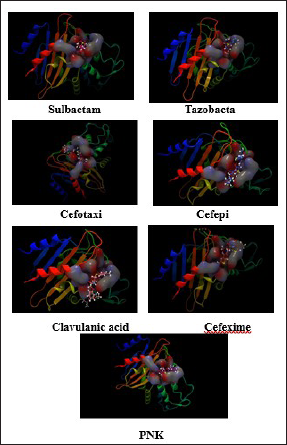
Docking result for the interaction of CTX-M-5 with ligands were shown in Table 4. The 3D diagrams depicting the interaction of CTX-M-5 with sulbactum, tazobactam, cefotacime, cefepime, clavulanic acid, cefexime, and PNK (from left to right) were shown in Figure 7.
CLC Score Toward the Interaction of Ligands with CTX-M-5.
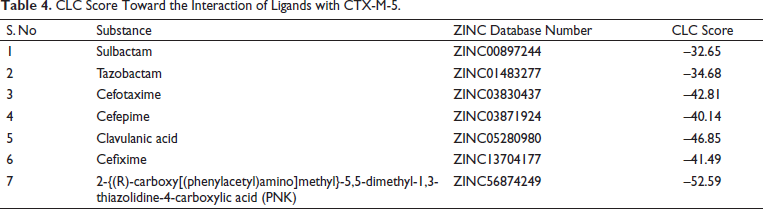
A β-lactam antibiotic when given in combination with a β-lactam inhibitor makes the antibiotic-resistant organism susceptible. As the inhibitor occupies the active site of the enzyme, the antibiotic is effective. The higher the negative energy, higher is “interaction energy,” contributing to efficiency. In our study, amongst drugs, cefepime showed higher interaction energy than cefotaxime and cefixime. On the other hand, among inhibitors, clavulanic acid with higher interaction energy proved to be an efficient inhibitor than tazobactam and sulbactam. It is noteworthy that PNK (D-benzyl penicilloic acid) molecule generated by the COACH server showed greater inhibition when compared to other molecules with higher interaction energy (Table 4). Previous studies reported that Benzylpenicillin has higher hydrolytic activity (kcat230 s−1) and affinity (K m 50 µM) to CTX-M-5 suggesting that our results are in coherence with their studies. So, PNK can be considered as strong inhibitor that can efficiently inhibit CTX-M-5.
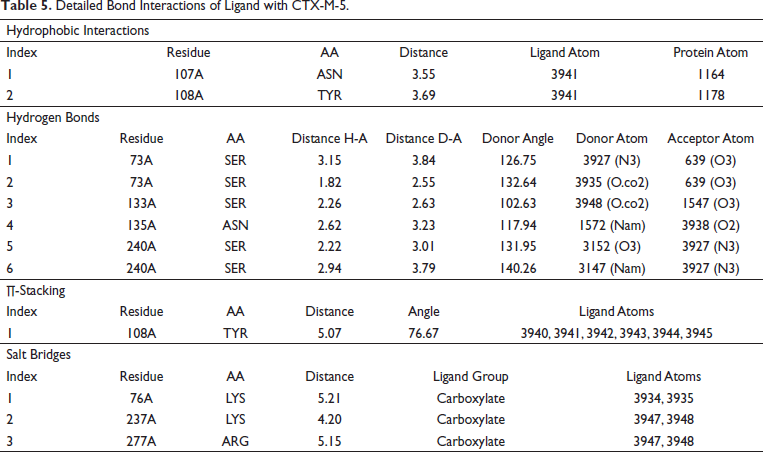
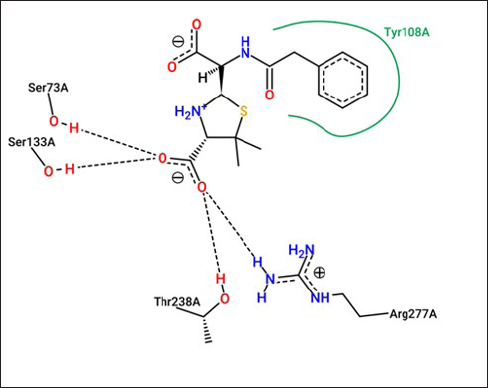
The 3D interactions of (PNK) ligand and CTX-M-5 were displayed by PoseView as shown in Figure 8. Protein-Ligand Interaction Profiler was used to obtain detailed bond interactions between the PNK and CTX-M-5 (Figure 9) and were tabulated Table 5). It was found that the amino acid residues Asn173, Ser240, Arg277, Gly239, Ser73, Thr238, Lys237, Thr219, Lys76, Ser133, Asn107, Glu169, Tyr108, and Asn135 enable correct positioning of the drugs and inhibitors within the active site of CTX-M-5.
. Detailed Bond Interactions of Ligand with CTX-M-5.
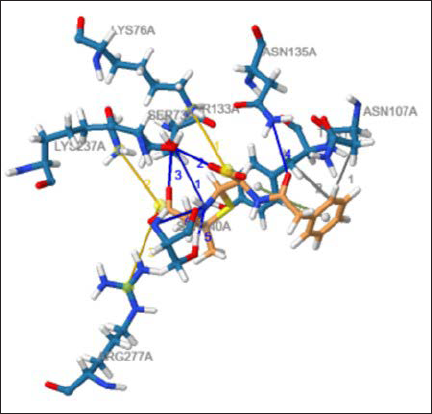
Discussion
Fluoroquinolones are commonly used antibiotics to treat invasive infections caused because of Salmonella. Misuse and overuse of fluoroquinolones led to antibiotic resistance. As a result, other antibiotics such as azithromycin and third-generation cephalosporins were used as the first-line therapy for treatment. 30 Increased cases of quinolone and cephalosporin resistant Salmonella serovars especially S. typhimurium in hospitals and communities are of great global concern in the management of Salmonella infections. The commonly reported extended-spectrum β-lactamases in different Salmonella serovars were majorly derived from SHV, TEM, and CTX-M type β-lactamase families.15, 31
CTX-M is highly prevalent among different serovars of Salmonella. CTX-M-5 one such plasmid-mediated ESBL belonging to CTX-M-2 group has been reported widely in S. typhimurium.10, 11, 32 In addition to CTX-M-5, the other members of the CTX-M-2 group include Toho-1, CTX-M-20, CTX-M-7, CTX-M-6, CTX-M-4L, CTX-M-4, CTX-M-2, CTX-M-65, and CTX-M-55.5, 33, 34 Intriguingly, CTX-M-5 is found only in human-associated S. typhimurium and confers a high level of resistance to cefotaxime. S. typhimurium causes gastroenteritis in humans and typhoid-like fever in a mouse model. Although the CTX-M-5, variant of CTX-M-2 was first reported in a gastroenteritis outbreak in children in Latvia in 1990, several nosocomial outbreaks involving CTX-M-5 ESBL S. typhimurium were reported in European countries such as Latvia (1991–1998), Russia (1996–1997), Hungary (1996–1998), Greece (1991–1998), Belarus (2007), the USA (2000, 2003, and 2007), and Russia, Belarus, and Kazakhstan (1994–2003). S. typhimurium is considered a zoonotic infection, i.e., it originates from animal sources, and, hence, it is assumed that transmission of CTX-M-type cephalosporinases/ESBL carrying S. typhimurium occurs from animals or animal products to humans via food chain. In contrast, CTX-M-5 is found only in human S. typhimurium isolates, and, hence, person-to-person transmission occurs. 15 Traditional β-lactamase inhibitors such as clavulanic acid, sulbactam, and tazobactam, as well as new inhibitors such as LN1-255 and NXL-104, reduce the activity of these enzymes.
Previously, various in silico studies on “CTX-M-drug” and “CTX-M-inhibitor” interactions were undertaken to identify the critical amino acid residues for enzyme interactions with medications and inhibitors. Shakil and Khan 19 used AutoDock for molecular modeling and docking of CTX-M15 with cefotaxime. They reported that the amino acids Asn 104,132, Gly 227, Thr 235, Gly 236, and Ser237 are important for positioning cefotaxime in the active site of CTX-M-15 and Ser237 and Arg276 residues in CTX-M-14 β-lactamase. 35 Mutational analysis and a high-resolution crystal structure of CTX-M-14 suggest that amino acid residues Ser237 and Arg276play a crucial role in catalytic specificity. 36 Docking study of CTX-M-9, 14, and 27 was done using Schrodinger by Kumar et al. 18 and found that the amino acids Asp 101, Asn 136, Lys 137, Asp 101, Glu 166, Ser 130, Asn 132, Thr 235, Ser 237, and Asp 240 are crucial for interaction with drug and inhibitor. Cefotaxime, cefixime drugs, tazobactam, and clavulanic acid inhibitors were shown to interact with the CTX-M-9 family of enzymes with higher energy. 18
In a similar molecular docking study done using Discovery Studio by Shakil and Khan, 35 using CTX-M variants (CTX-M-15, CTX-M-53, CTX-M-71, CTX-M-82, and CTX-M-89), it was observed that ceftazidime drug interacted with the amino acid residues of L/Y291, G290, D289, T288, R285, W232, S231, A/T230, P229, L228, G227, and A226 of CTX-M-type 2009 variants. Additionally, among the different inhibitors studied, sulbactam was binding most efficiently with high interaction energy. 19
Bethel et al. 37 studied the inhibition of CTX-M-9 by sulbactam, tazobactam, clavulanate, meropenem, doripenem, ertapenem, and 6-methylidene penem using electrospray ionization mass spectrometry. They reported that the amino acid residue Arg276 of the CTX-M-9 contributes to the region of positive charge near the catalytic site, which is very essential for CTX-M-enzyme activity. More specifically, c3 carboxylase inhibitors are recognized by the Arg276 amino acid residue of CTX-M-9. 37 Danishuddin and Khan 38 modeled and studied the docking of PCR amplified CTX-M β-lactamase with traditional inhibitors such as sulbactam, clavulanic acid, and tazobactam followed by novel β-lactamase-inhibitors such as LN1-255 and NXL-104 using the software, GOLD 5.0. Among the tested inhibitors, compound LN1-255, and tazobactam have shown to be the best inhibitor against the enzymes CTX-M-15, SME-1, and IMI-1. Amino acid residues Gly237, Thr236, Lys235, Ser130, and Ser70 were reported to be essential for the formation of enzyme-inhibitor complexes by hydrophobic interactions and hydrogen bonding. 38 Ali et al. 39 have studied the interaction of non-β-lactam inhibitors against the CTX-M-15-type β-lactamases through a virtual screening protocol. Three non-β-lactam inhibitors such as RF04517, BTB04243, and RJC01340 interacted with CTX-M-15 very tightly and interaction is stabilized through hydrogen bond and hydrophobic interaction by Asn104, Asn132, Ser237, and Asp240 amino acid residues. Ser237 creates a strong hydrogen connection with the acyl-amide cefotaxime chain. These compounds were experimentally validated against E. coli strain carrying CTX-M-15 β-lactamase. 39 Furthermore, Maryam et al. 40 showed the importance of amino acid residues Asn-247 and Arg-64 located in the proximity of the catalytic site of the CTX-M-15-type β-lactamase by site-directed mutagenesis. E. coli cells expressing Asn247Val and Arg64Leu mutant CTX-M-15-type β-lactamases have shown reduced cefotaxime minimum inhibitory concentrations of 512- and 128-fold, respectively. Similarly, in in vitro kinetic studies, the catalytic efficiencies of the CTX-M-15 (N247V) and CTX-M-15 (R64L) mutant β-lactamases were reduced by 89.66% and 71.11%, respectively. 38
Conclusion
In the current study, CTX-M-5 of S. typhimurium was modeled and validated using 3D structure validation tools. Besides this, conserved sites and the ligand-binding sites were also determined to predict the active site of the protein. Of the drugs and inhibitors used, Cefipime and clavulanic acid were found with higher interaction energies and effective. Above all, the COACH server predicted that PNK (D-benzylpenicilloic acid) molecule competently inhibited CTX-M-5 with greater negative energy. Previous studies reported that the PNK molecule, which is a diastereomer of benzylpenicllinis high affinity compound. So, we infer that our docking results are compatible with the wet lab studies. We also identified the amino acid residues of the active site responsible for the positioning of drugs and inhibitors. With the information above, alterations in the amino acid residues could aid to develop more potent and adaptable inhibitors. This is the first time to report an in silico modeled 3D structure of CTX-M-5, conserved sites of CTX-M-5, and all the commonly found amino acid residues that are crucial in hydrogen bonding interactions. These conserved sites can be further used in the rational design of novel drugs and inhibitors.
Footnotes
Acknowledgment
We acknowledge the technical help of Dr Sathish Ganji for the critical appraisal of the article. Dr Uday Sankar Allam gratefully acknowledges the Department of Science and Technology (DST) for their support through the Early Career Research Award and DST-FIST. Mohammad Shaik Jasmine acknowledges University Grants Commission, India for Maulana Azad National Fellowship.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Statement
Ethical approval is not applicable for this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
There are no human subjects in this article and informed consent is not applicable.