
Abstract
Background:
Hypomyelinating leukodystrophies (HLDs) are rare, progressive, autosomal recessive disorders that impair the central nervous system (CNS). The PYCR2 (Pyrroline-5-carboxylate reductase 2) gene mutations cause HLD type 10, leading to microcephaly, hypotonia, developmental delay and intellectual disability.
Case Summary:
We report a four-year-old boy from Karnataka, India, born to a second-degree consanguineous union, presenting with global developmental delay, progressive microcephaly, hypotonia and limb weakness, where the condition was categorised as cerebral palsy (CP). Whole-exome sequencing (WES) identified a homozygous PYCR2 variant (NM_013328:c.751C>T; p.Arg251Cys), confirmed by Sanger sequencing, with heterozygosity in both parents and the elder sibling. This study represents the third reported case in India.
Conclusion:
This study highlights the importance of a molecular diagnostic approach for CP mimics. Identification of mutations enables accurate diagnosis, informed genetic counselling and early interventions, as well as recurrence prevention, emphasising the need for awareness of rare HLDs in clinical practice.
Genetic testing plays a crucial role in differentiating cerebral palsy (CP)-like symptoms from rare conditions such as PYCR2-related HLD10, allowing for precise diagnosis and appropriate genetic counselling for families.Key Message:
Introduction
Hypomyelinating leukodystrophies (HLD) are very rare, progressive, clinically overlapping and genetically heterogeneous encephalopathies with autosomal recessive inheritance that impair myelin formation in the central nervous system (CNS).[1] Affected individuals typically exhibit progressive microcephaly, hypotonia, spasticity, global developmental delay, intellectual disability, failure to thrive, seizures and cerebral hypomyelination.[2] Mutations in the Pyrroline-5-carboxylate reductase 2 (PYCR2) gene [OMIM #616406] have been reported in patients from consanguineous families with hypomyelinating leukodystrophy 10 (HLD10).[2] The PYCR2 gene encodes the mitochondrial enzyme pyrroline-5-carboxylate reductase 2, which converts to proline, which is an essential amino acid for the CNS and connective tissue function. A defect in proline synthesis leads to significant impairments, including intellectual disability and hypomyelination.[3] In this study, we describe a PYCR2 gene mutation that is linked to hypomyelinating leukodystrophy type 10 in a patient presented with cerebral palsy (CP)–like features.
Case Report
The patient was a four-year-old boy from a town in Karnataka, India. His birth history revealed a normal prenatal period and a normal vaginal delivery, with a birth weight of 2,500 g. The parents were related by a second-degree consanguineous marriage. He has an elder sister, who is developmentally normal and there was no family history of hereditary, neurological, psychological or congenital disorders; the mother reported a history of miscarriage. He had a fever two days after birth. Since birth, he had poor motor functions with upper and lower limb weakness. He started to babble at about 1.2 years. As he grew older, poor mental ability, limited speech and language and restricted tongue movements were observed. Both hearing and vision assessments were reported as normal. The child exhibited poor vegetative functions and was diagnosed with global developmental delay. The medical history states that the patient was diagnosed with the disease after the Magnetic Resonance Imaging (MRI) revealed symmetrical white matter demyelination with associated atrophic signal changes, suggestive of a metabolic or genetic leukodystrophy. Features were doubtful for a genetic condition in view of progressive microcephaly, ataxia and intellectual disability.
He was brought to the All India Institute of Speech and Hearing (AIISH) for speech assessment, therapy and follow-ups. As part of the intramural CP project undertaken by the Unit for Human Genetics at AIISH, blood samples were collected from the family for genetic studies. DNA was isolated from blood tissue by PureLink™ Genomic DNA Mini Kit (Thermo Fisher Scientific, USA). The sample was subjected to whole-exome sequencing (WES) using the Ion Proton sequencing platform (Life Technologies, USA). The sequencing data achieved an average coverage depth of 80.1×, with 98.9% of reads successfully aligned to the reference genome. Variant calling was performed directly from the raw sequencing data using the built-in variant caller plugin in Torrent Suite (version 5.2.2). A total of 38,668 variants were identified and subsequently annotated against the human reference genome assembly GRCh37 (hg19) using Ion Reporter™ software (version 5.18) (Thermo Fisher Scientific, USA).
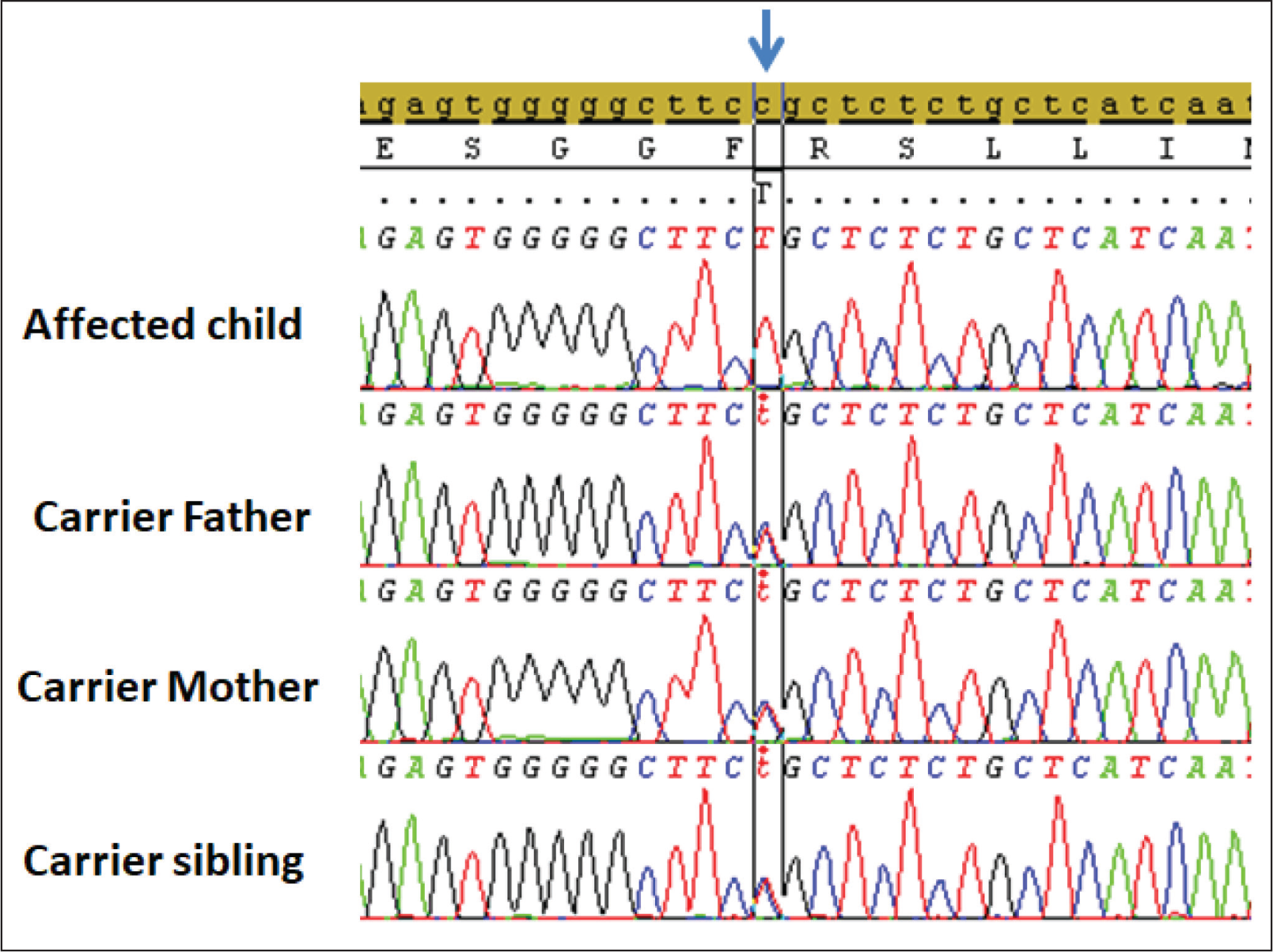
According to the WES results, a homozygous variant PYCR2: NM_013328:c.751C>T (p.Arg251Cys), associated with HDL10, was identified in the proband (Figure 1). The variant resulted in a missense variant in PYCR2, leading to the loss of normal protein function. The identified variant was classified as pathogenic based on the ACMG (American College of Medical Genetics and Genomics) criteria (PM1, PP2, PP3, PP5), using the Franklin bioinformatics platform (
Schematic illustration of the Sanger sequencing results of the affected child and unaffected family members. The affected child shows a homozygous PYCR2 mutation (NM_013328:c.751C>T [p.Arg251Cys]), as indicated by the arrow. The other sequences show a mixture of C and T, signifying the presence of a heterozygous variant. Positions of variants are according to RefSeq KU178615.1 (cDNA)
Discussion
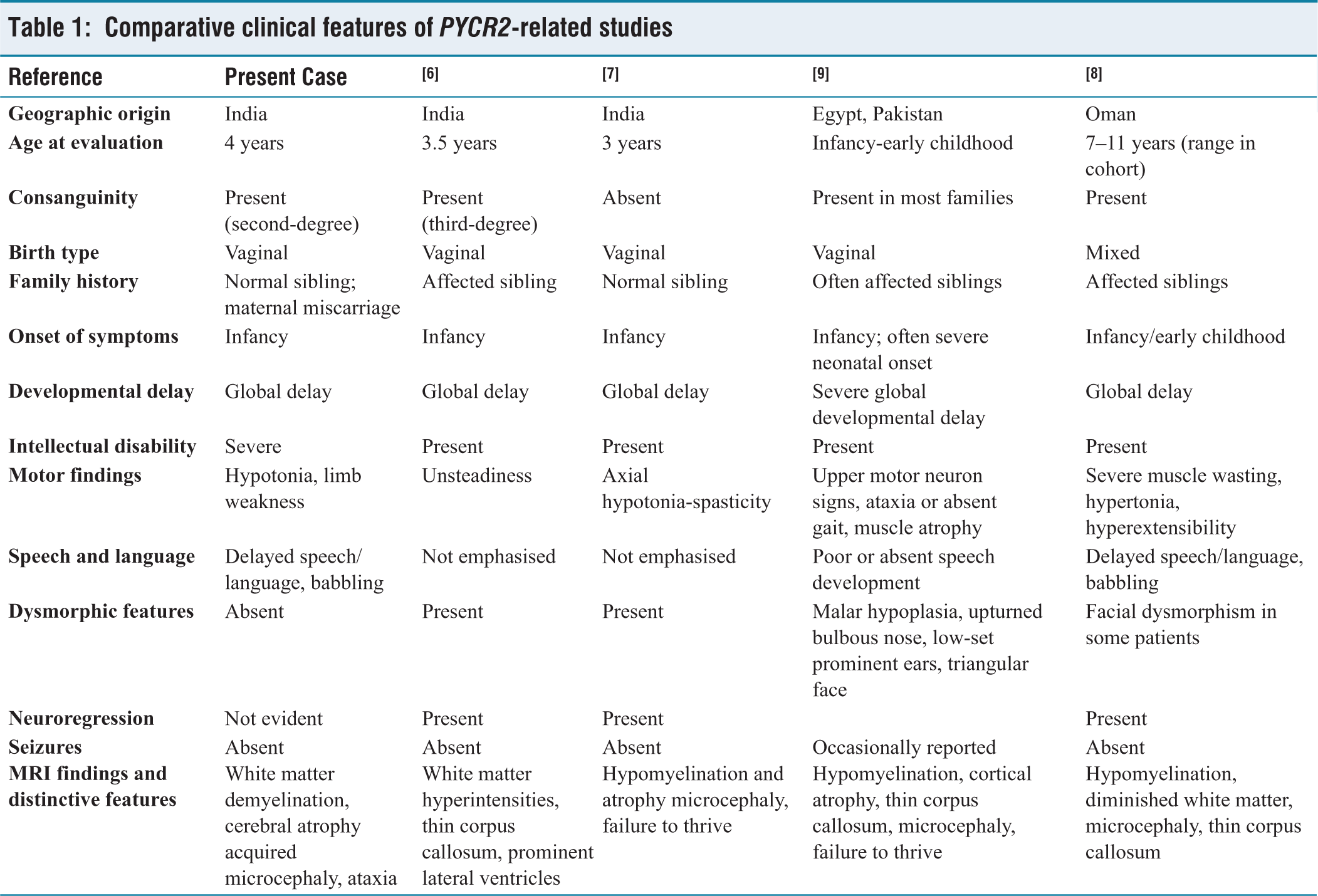
The classification of a patient under CP is primarily based on the clinical recognition of characteristic features. However, a range of metabolic, neurodegenerative and developmental genetic disorders can present with overlapping manifestations that resemble the CP phenotype.[4] As demonstrated in this case, the patient showed similar indications. However, after the genetic testing, it was revealed that he carried the PYCR2: NM_013328:c.751C>T mutation that lies in the dimerisation domain of the PYCR2 protein, which causes HLDs.[8] The clinical presentation was consistent with the genotype, supporting the diagnosis. HLDs represent a clinically and genetically heterogeneous group of neurodevelopmental disorders characterised by defective or incomplete myelin formation in the CNS and only 38 cases have been reported so far.[5] The clinical features observed in our patient, including pre- and postnatal growth deficiency, progressive microcephaly, psychomotor impairment, intellectual disability and muscle weakness, are consistent with previously reported cases [Table 1]. Previous studies have reported only two cases of PYCR2-related HLD10 from India. One study identified the PYCR2 c.751C>T (p.Arg251Cys) variant,[6] the other study reported the PYCR2 c.356G>A (p.Arg119His) variant.[7] To the best of our knowledge, this study represents the third reported case of PYCR2-related HLD10 from India.
Comparative clinical features of PYCR2-related studies
HLD10 is inherited in an autosomal recessive manner. The patient’s parents were heterozygous carriers of the PYCR2:c.751C>T variant [Figure 1]. Owing to their consanguineous union, the patient inherited two mutant alleles, resulting in HLD10, while the elder sibling was found to be a heterozygous carrier. Studies have reported patients from consanguineous and non-consanguineous families who carried a homozygous PYCR2 mutation and exhibited similar clinical features.[2,7–9] Patients with HLD10 have a high mortality rate, with most affected children not surviving beyond 10 years of age.[10] These findings underscore the clinical progression of HLD10 and highlight the need for continued investigation into its underlying pathophysiological mechanisms.
In conclusion, we presented a patient with homozygous missense mutations in PYCR2 as the underlying cause of HLD10, characterised by developmental delay, microcephaly and hypomyelination. This case highlights the importance of adopting a genotype-based approach for diagnosis that clinically resembles CP. Nevertheless, further studies are required to better understand the underlying disease mechanisms and to facilitate earlier detection and treatment.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Institutional ethical committee approval number
The Ethics Committee of the AIISH approved the study in accordance with local guidelines (approval number SH/CDN/ARF-40/2016-17).
Informed consent
Informed consent was obtained from the patients for the publication.
Credit author statement
NS, SN: Conception, supervision, data acquisition.
SM, JT: Supervision, data acquisition.
CS: Data analysis, manuscript writing.
Data availability
Data will be shared upon request.
Use of artificial intelligence
None.