
Abstract
Tremor is a heterogeneous symptom and may be caused by genetic and non-genetic causes. Tremors in the paediatric age group associated with other features may be suggestive of a syndromic diagnosis. We describe the case of a 13-year-old boy who presented to the neurology department with progressive tremors. A reflex genetic evaluation revealed mild-moderate intellectual disability, subtle dysmorphic features and action-induced coarse tremors. A high-resolution chromosomal microarray confirmed the presence of 9p13.3 microdeletion syndrome. We delineate the core features of this syndrome and provide a comparison of all the reported features associated with this contiguous deletion syndrome.
Introduction
Tremor can be a disabling symptom and is etiologically heterogeneous. Most tremors of childhood can result from a neuro-developmental/neuro-metabolic/neuro-degenerative condition or may be syndromic, when associated with other symptoms. We report a young Indian boy who presented with progressive tremors. Upon evaluation, he was found to have subtle dysmorphic features and a mild intellectual deficit, which eventually led to the discovery of a rare microdeletion syndrome. This is probably the first report of 9p13.3 microdeletion syndrome from India.
Case History
A 13-year-old boy was brought by his parents to the neurology clinic with the chief complaint of recent onset tremors. The attending neurologist also uncovered mild-moderate intellectual disability, introverted behaviour and facial features, which were mostly attributed to perinatal asphyxia. The boy was referred to the genetic clinic for further evaluation, since the brain imaging did not reveal any changes consistent with hypoxic ischaemic sequelae. He was the first offspring of non-consanguineous parents, born via a preterm caesarean section at 8 months, in view of oligoamnios. There were no maternal illnesses, except for mild pedal oedema, which was conservatively managed. Scans revealed mild intrauterine growth restriction and oligoamnios from sixth month onwards. His birth weight was 2.2 kg and his birth cry was delayed. He was observed in the neonatal intensive care unit for 3 days and then discharged. Breastfeeding was normal. Parents report mild global delay with walking and bisyllables attained by 2 years. He was also noted to have intellectual disability with poor scholastic performance.
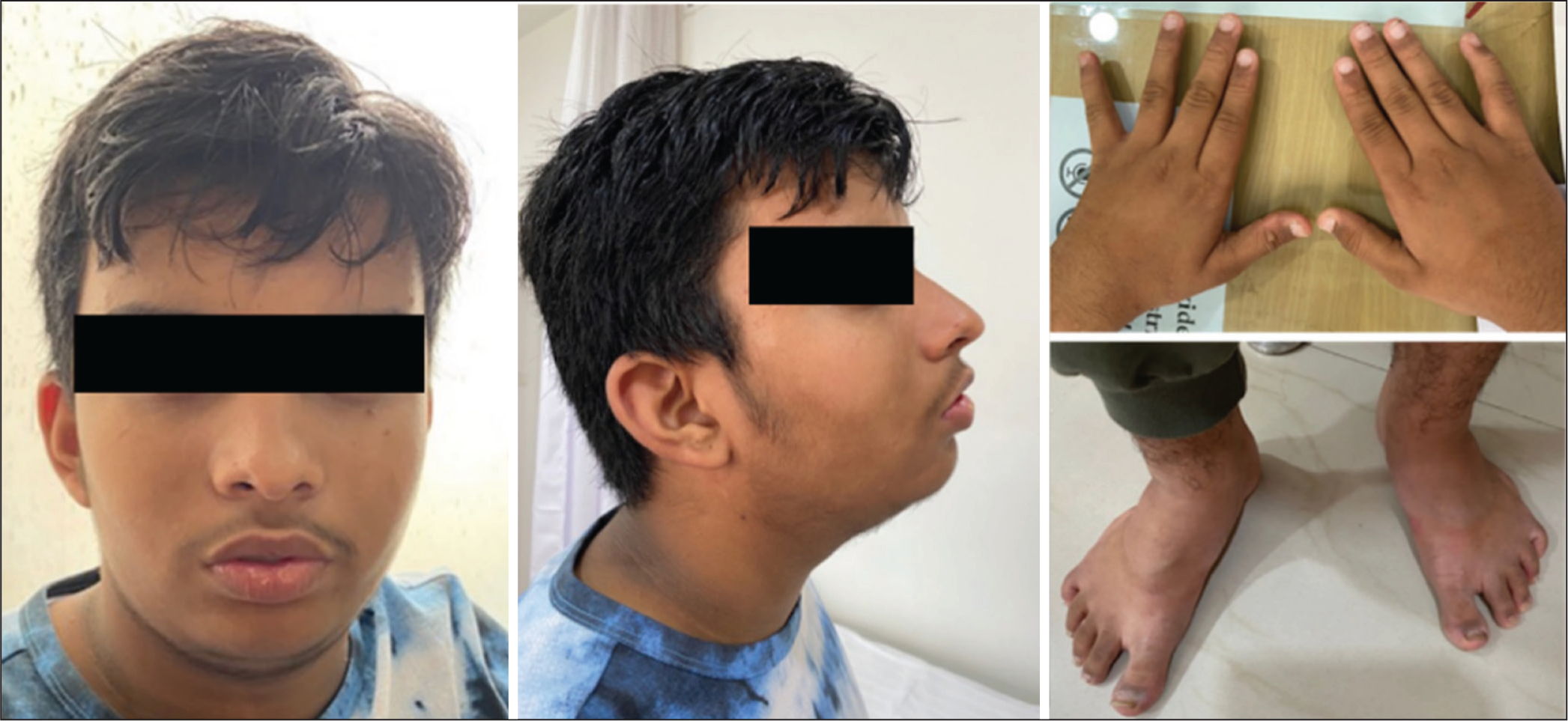
On examination, he was alert, responded briefly to queries, but was cooperative. His head circumference, height and weight were at 70th, 65th and 95th centiles, respectively. Dysmorphism was noted in the form of dolichocephaly, oval face, thick bushy, flared eyebrows, bilateral prominent anteverted ears, retrognathia, low-set ears, bilateral gynaecomastia with inverted, downturned nipples, increased sandal gap, excessive skin striae and acanthosis nigricans. The clinical photographs of the patient have been included in Figure 1, with informed consent from the family. Neurological examination revealed suboptimal higher mental functions, slightly slurred speech and normal muscle tone with preserved power and reflexes. The tremors were hyperkinetic, more obvious with action and sustained throughout the task.
Dysmorphic features noted in the proband
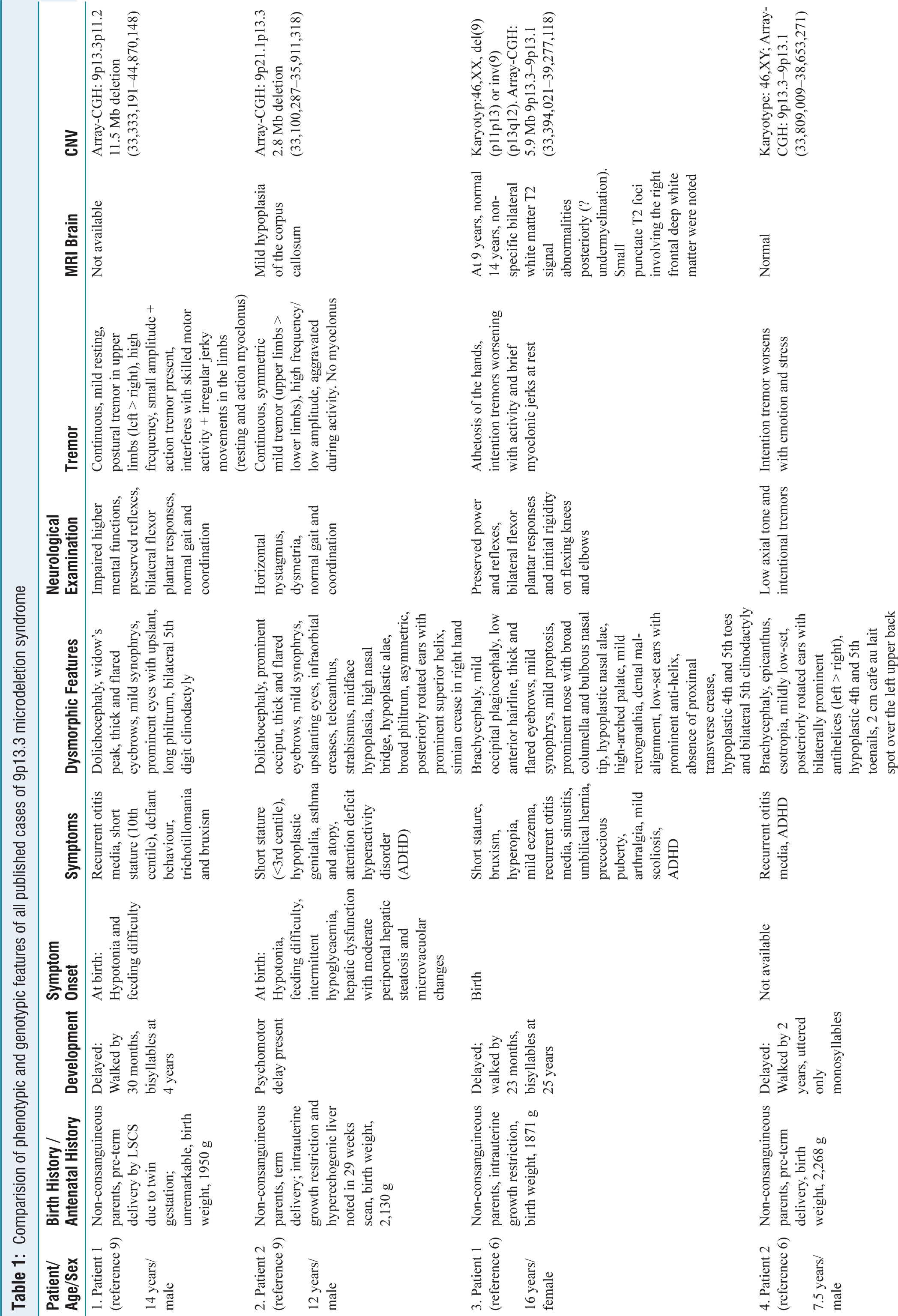
Blood investigations, including thyroid profile, were normal. MRI brain and EEG were unremarkable. Intelligence quotient is 46, corresponding to moderate intellectual disability. The triplet primed polymerase chain reaction for fragile X syndrome was normal. Exome sequencing implicated the presence of a heterozygous deletion involving chromosome 9. Chromosomal microarray was performed using an Illumina Bead Array technology, consisting of 750K genome-wide oligonucleotide probes and the analysis is based on the Human reference genome version GRCh38. It revealed a heterozygous, pathogenic 5.3 Mb deletion on the short arm of chromosome 9 between the breakpoints 33408036–38771111, indicating monosomy for the region 9p13.3. The identified deletion overlaps with the region reported in the 9p13.3 deletion syndrome. This region contains 72 OMIM genes, 21 of them being morbid [Figure 2B]. Haplo-insufficient genes include PAX5, VCP, AQP3, NPR2, TPM2, RUSC2 and UBAP1 [Figure 2A]. Many deletions have been previously reported in this region, with several overlapping larger and smaller deletions [Figure 3]. Parent samples were unavailable for segregation analysis. The phenotypic and genotypic features of all published cases of 9p13.3 microdeletion syndrome have been compared in Table 1.
Comparision of phenotypic and genotypic features of all published cases of 9p13.3 microdeletion syndrome
(A) OMIM genes, and (B) Morbid genes encompassed within the breakpoints GRCh37:2:236309675-242099260 in our patient
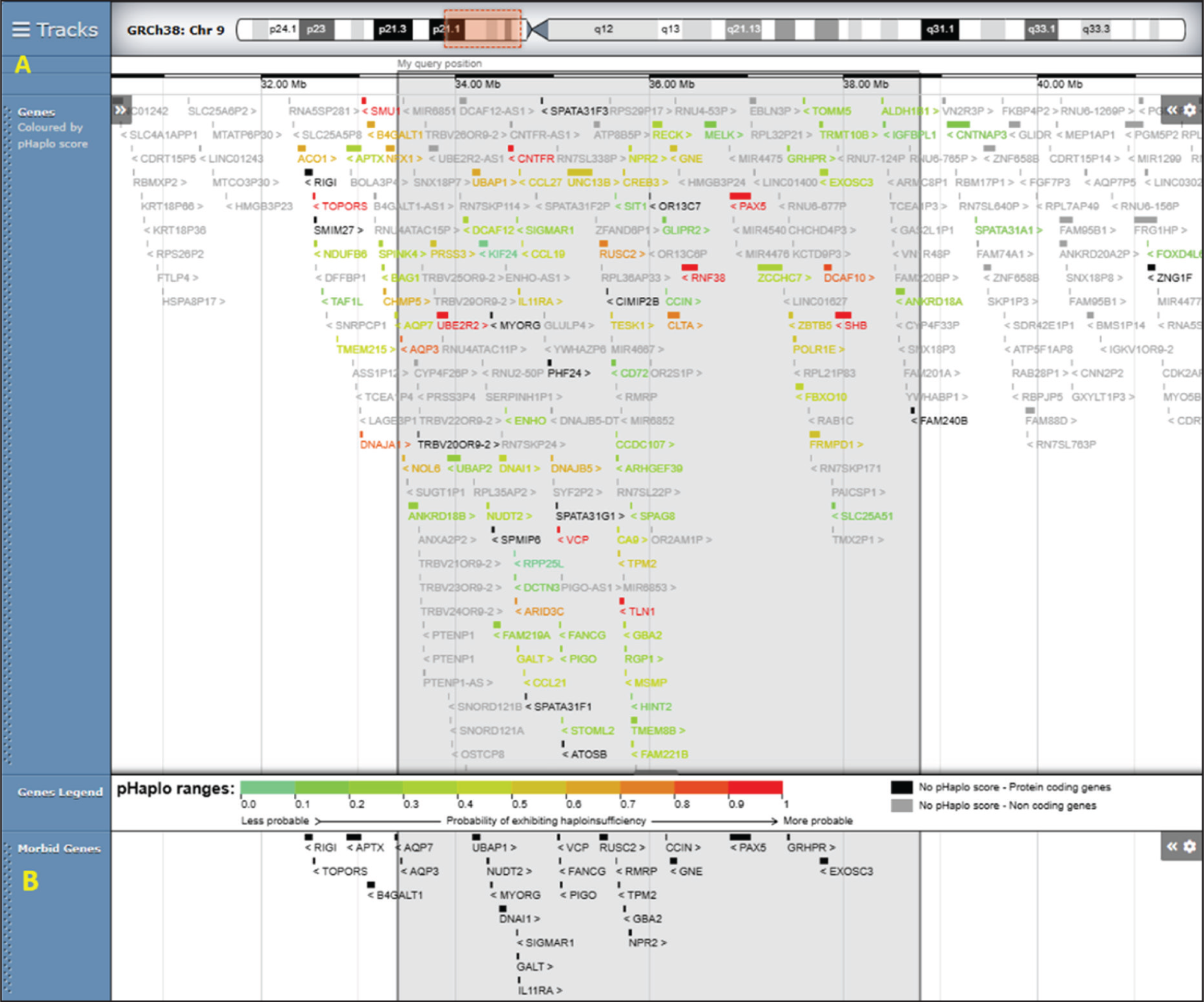
Breakpoints of patients with 9p13.3 microdeletions reported previously on DECIPHER genome mapping tool GRCh38 v11.30. The blue box highlights the DECIPHER IDs of the patients with the closest breakpoints to our patient

Discussion
9p minus syndrome or Alfi syndrome (OMIM# 158170) was first reported in 1973, with a recognisable set of features in six patients.[1] In 2008, 10 patients were described with distal deletion of 9p with features such as dysmorphism, trigonocephaly, intellectual disability, impaired speech, cardiac and genital abnormalities.[2] The deletion sizes ranged from 800 kb to 12.4 Mb, with minimal phenotypic variability across the deletion sizes. The largest group of 9p- was studied, comprising data from 719 patients; 35%–40% of breakpoints occurred between 9p22–9p24, while the least (0.6%) occurred between 9p11–9p12.[3]
Our patient has a 9p deletion with a proximally placed breakpoint that is, at 9p13. The first microscopically visible 9p13 syndrome was reported way back in 1994 in a female child with dysmorphism, abnormal genitalia and feeding difficulties.[4] Another 9-year-old girl with additional features such as craniosynostosis and attention deficit hyperactivity has been described.[5] With the advent of chromosomal microarray, many more cases of 9p13.3 syndrome were subsequently reported, notably a report of two unrelated young patients, a 9-year-old girl and a 7.5-year-old boy and another study of tremors in a child as young as 3 years.[6,7] A deletion with breakpoints very similar to that of our patient was recently described in a female fetus with omphalocele, limb defects such as metacarpal agenesis and symbrachydactyly. Autopsy evaluation had also revealed other features of 9p, such as trigonocephaly, dysmorphism and sex reversal.[8]
Most recently, two unrelated patients with 9p13.3 syndrome have been described and recognised tremor as a major feature of this distinct condition. They also discovered a 2 Mb region encompassing 70 OMIM genes that is shared among 9p13.3 patients and postulated that the NPR2 gene may be responsible for the core phenotype.[9] Our patient shared many cranio-facial features already described, abnormal skull shape, developmental delay, dysmorphism, mild intellectual disability and tremors. However, he was mostly introverted, calm and composed as opposed to previous reports of aggression, social or hyperactive personality. He was largely independent for daily activities and interacted amicably with peers and elders. A few of the commonly reported features that were absent in our patient include trigonocephaly, short stature, social personality or precocious puberty. At the time of examination, he was in the peri-pubertal phase with some facial hair, but other pubertal features were evolving. The genitalia were normal.
The haploinsufficient genes encompassed within our patient’s breakpoints include PAX5, VCP, AQP3, NPR2, TPM2 and UBAP1. Among these, NPR2, UBAP1, TPM2, and VCP cause morbid phenotypes. UBAP1 is associated with Spastic paraplegia 80, TPM2 causes arthrogryposis, congenital myopathy, and VCP is associated with 3 phenotypes, namely – Charcot Marie Tooth disease 2Y, Fronto-temporal dementia, and inclusion body myopathy with early onset Paget disease. NPR2 is known to cause short stature, skeletal dysplasia and more recently, associated with the core phenotype of the 9p syndrome.
We attempted a genotype-phenotype correlation with the reported cases of 9p13.3 syndrome via DECIPHER version 11.36, Genome mapping Tool. There are 3 other reported deletions with closely overlapping breakpoints as our patient - DECIPHER ID: 503405, 385177, 472489 as shown in Figure 3. Patient ID 503405 has a 5.74 Mb de novo deletion, with distal breakpoint extending slightly beyond our patient’s breakpoint. The phenotype comprised of global delay, severe speech, microcephaly, postnatal growth deficits, intraventricular hemorrhage, anomalies like strabismus, bifid uvula, hyperdistensibility at elbow, and finger flexion contractures. Patient ID 385177 has a 5.32 Mb deletion, with proximal and distal breakpoints aligning exactly as our patient. However, the only reported phenotype is sleep impairment and athetosis. Patient ID 472489 has a deletion 5.81 Mb with breakpoints extending slightly beyond in both directions. The reported phenotype includes developmental delay, speech impairment, intellectual disability, thin upper lip vermillon strabismus, hypermetropia, sleep difficulties, and seizures.
Our patient represents yet another discernible 9p13.3 phenotype, mild developmental delay, intellectual disability, subtle dysmorphism and tremors, without other associated anomalies. We emphasise that genetic evaluation is important in young patients presenting with tremors or other neurological features.
Conclusion
The 9p13.3 microdeletion syndrome is a distinct genetic condition with highly variable features attributable to the variable deletion sizes, and penetrance. Core phenotype includes craniofacial dysmorphism, intellectual disability, short stature and behavioural abnormalities. Tremor is a chief symptom and may have a variable age of onset, ranging between infancy and adolescence. Most of the reported tremors are hyperkinetic and action induced. Haplo-insufficiency appears to be the most important pathogenetic mechanism underlying the core phenotype. 9p13.3 syndrome is one of the few microdeletion syndromes with tremor as the predominant symptom.
Footnotes
Acknowledgements
We thank the patient and his family for their complete cooperation. We also thank MedGenome Labs, Private Limited, for performing the diagnostic genetic testing for this patient.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Institutional ethical committee approval number
Not applicable.
Informed consent
Written informed consent was taken.
Credit author statement
We hereby confirm that the manuscript titled ‘9p13.3 syndrome: A rare and unusual genetic cause of a tremor in a young Indian boy’ has been read and approved by all the authors, that the requirements for authorship have been met and this article reflects genuine work by each one of us.
Data availability
Not applicable.
Use of artificial intelligence
None.