
Abstract
Background and Aims:
Interdigitating dendritic cell tumour/sarcoma (IDCS) is an exceedingly uncommon and aggressive tumour with a dismal prognosis. IDCS arises from antigen-presenting cells, which involve nodal and extranodal sites. The aim of the study is to evaluate a case of IDCS and its diagnostic challenges.
Methods:
A 21-year-old female with no comorbidities, a bone marrow aspirate and biopsy and an excision biopsy of the lymph node shows atypical mononuclear infiltrates that were immunohistochemically positive for S100-diffuse, CD68, LCA and Ki67.
Results:
Here, we report a single case of IDCS with a tumour arising from the lymph node and bone marrow infiltration.
Discussion:
IDCS is an aggressive disease with uncertain behaviour; strong suspicion is required to establish a diagnosis.
Introduction
Non-lymphoid and non-phagocytic antigen-presenting dendritic cells are engaged in T-cell priming and maintain the anatomical and functional integrity of lymphoid organs. Dendritic cells (DCs) are specialised professional antigen-presenting cells that are essential to the cell-mediated adaptive immune response against foreign antigens because they activate T-cells.[1] DCs can be further divided into resident and migrating DCs within the lymph node. Follicular dendritic cells (FDCs) and interdigitating dendritic cells (IDCs) are two types of resident DCs that reside in lymphoid organs. In contrast to FDCs, IDCs are produced from bone marrow–derived hematopoietic stem cells. IDCs are present in T cell–rich regions of peripheral lymphoid tissue, such as the tonsils and lymph nodes’ paracortex and deep cortex, the splenic periarteriolar lymphoid sheaths and the interfollicular regions of mucosa-associated lymphoid tissue (MALT). IDCs are immunoreactive with S-100 and resemble Langerhans cells in light microscopy appearance; however, they do not feature ultrastructural Birbeck granules or constant CD1 expression. They are distinguished by weak staining for ATPase, alkaline phosphatase and nonspecific esterase. As with FDCs, IDCs express a large number of macrophage receptors, but they do not complement.[2] Dendritic cell neoplasms are exceedingly uncommon and aggressive, with morphological and phenotypical overlap and heterogenicity. In 1981, the first case in the world was reported by Feltkamp and coworkers.[3] In 2002, the first case in China was reported by Wei-Dun Xu et al.[4] Concerning the sites, nodal and extranodal involvement can occur. Extranodal sites include the liver most commonly, followed by the gastrointestinal tract, lung, and spleen,[5] nasopharynx, mesentery and oral cavity. IDCS shares morphological features of both lymphoma and sarcoma; henceforth, diagnosis is established by cytomorphological and immunohistochemical features.[6]
Clinical Presentation
A 21-year-old female with no comorbidities complained of urticarial rashes all over the body for one week, and simultaneously she developed high-grade fever, dry cough, exertional dyspnea and sudden-onset bilateral progressive leg swelling in the past one week with no diurnal variation. A history of significant weight loss and loss of appetite was present. Per abdomen, hepatosplenomegaly was present. Peripheral smear examination revealed normocytic normochromic anaemia, leucopenia and thrombocytopenia with an increased reticulocyte count. Smears for haemoparasites were negative. Elevated levels of ferritin, triglycerides, alkaline phosphatase and LDH were found as well as decreased albumin levels. PET-CT showed hepatosplenomegaly with diffuse metabolic activity and mildly increased uptake in lymph nodes (cervical, abdominal and mediastinal regions).
Clinical diagnosis revealed pancytopenia with hepatosplenomegaly for evaluation, to exclude haematolymphoid neoplasms. Further plans include a cervical lymph node excision biopsy, a bone marrow aspirate and a bone marrow biopsy.
Results
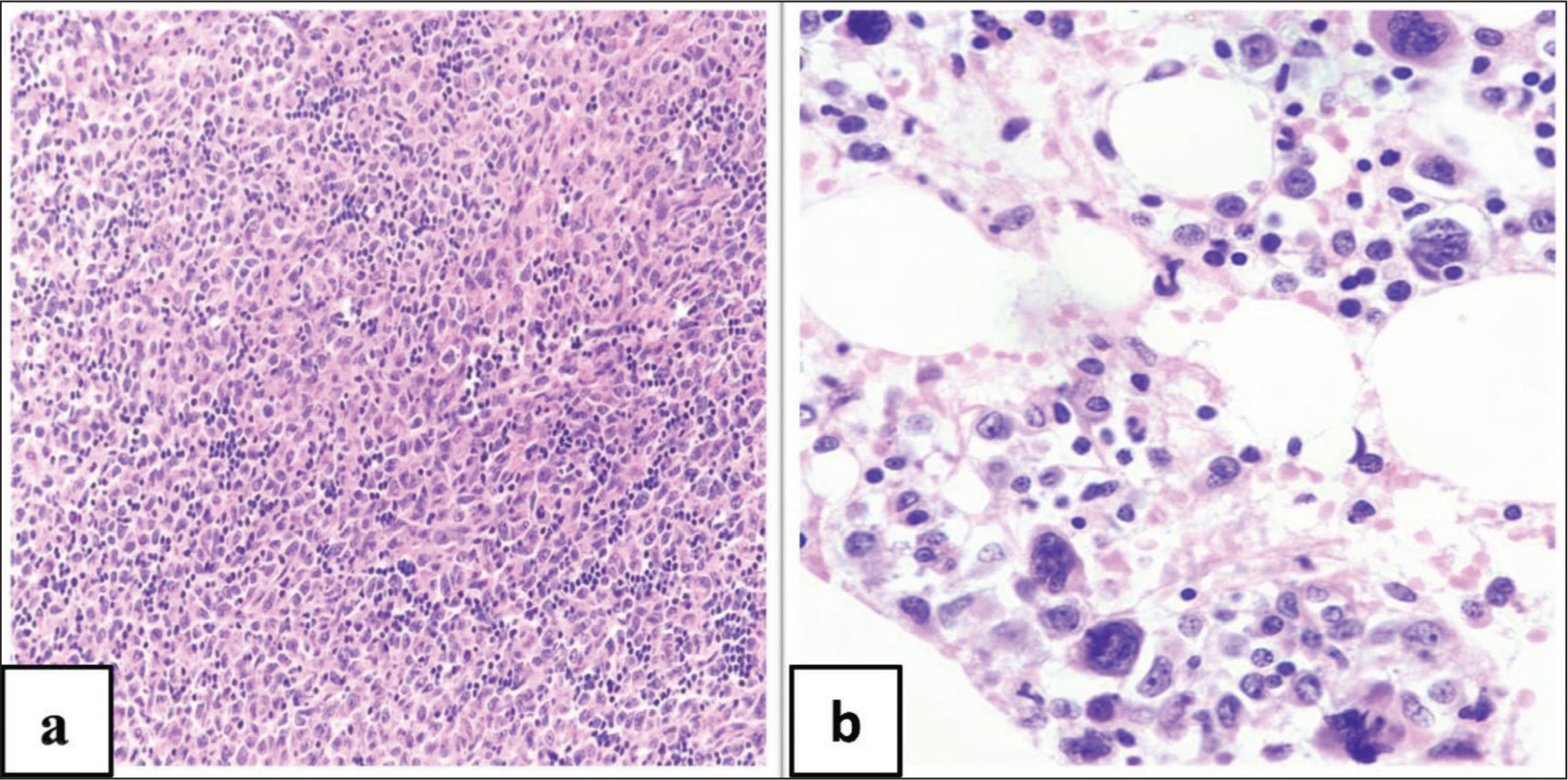
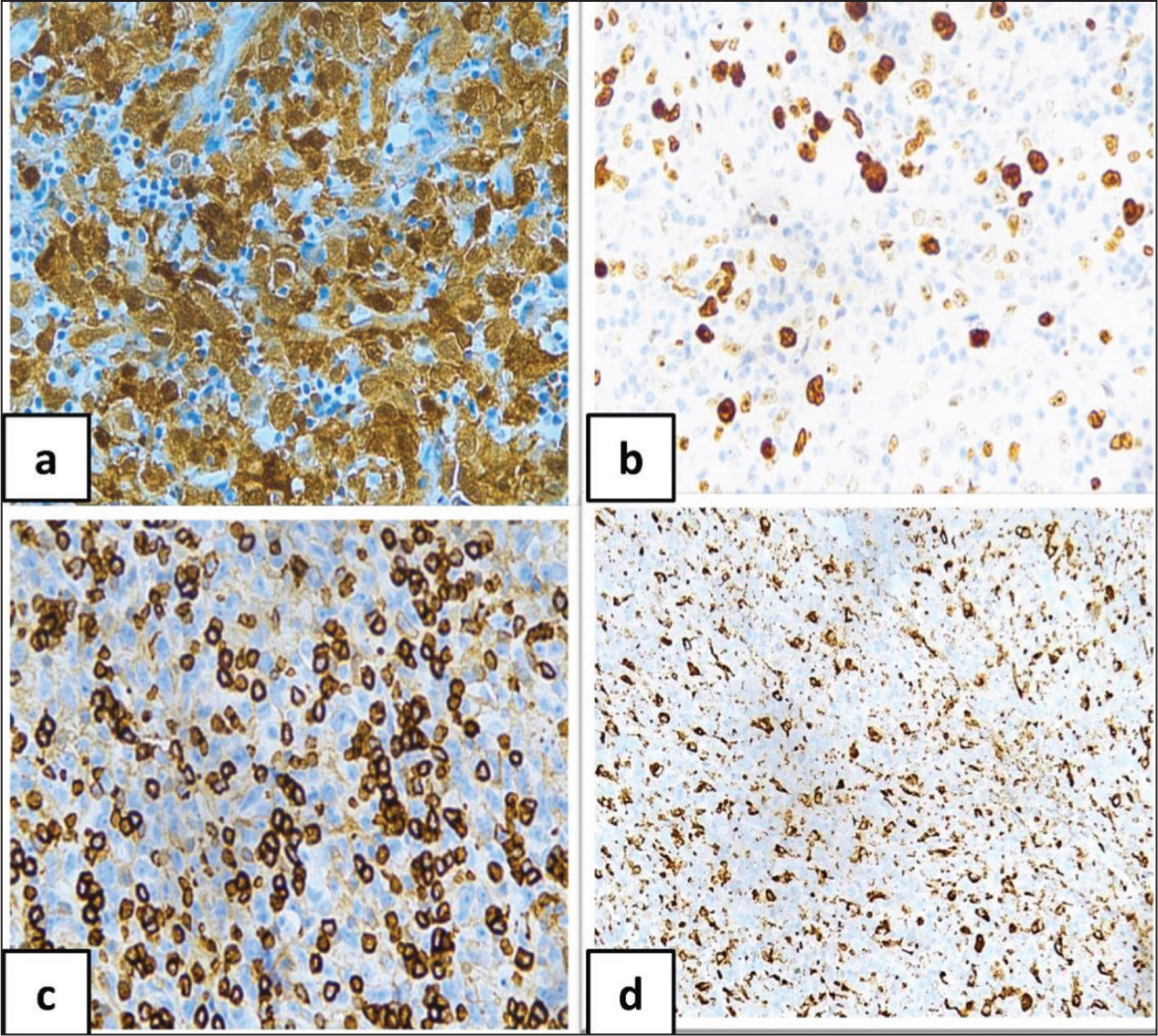
Histopathological examination of excision biopsy of cervical lymph node shows an effaced architecture with paracortical expansion of atypical cells. They are plump spindle cells arranged in fasicles and sheets with mitotic figures, as observed in an excision biopsy of a lymph node. The bone marrow aspirate and biopsy shows an atypical mononuclear infiltrate (Figure 1). Immunohistochemically interdigitating cytoplasmic processes show S100: strong and diffuse positive, Ki67: high proliferative index; leucocyte common antigen (LCA): positive, CD68: granular cytoplasmic pattern (Figure 2), CD4: positive, CD1a and Langerin: negative markers of Langerhans cell histiocytosis, FDC markers CD21 and CD23: negative, and CD20 B-cell marker: negative. The melanocytic marker HMB-45 was negative. CD138, CD30, CD43, Myeloperoxidase and CD117 were also negative.
(a) H&E 400X Large Cells with Prominent Nucleoli, Abundant Eosinophilic Cytoplasm, Background Shows Lymphocytes and Plasma Cells. (b) H&E 400X Shows Atypical Mononuclear Infiltrates
The Neoplastic Cells Were Positive for Following Immunohistochemical Stains: (a) 400X S100; (b) Ki67 400X High Proliferative Index; (c) CD45 (Leucocyte Common Antigen) 400X; (d) CD68 400X
Discussion
IDC develops from hematopoietic precursors via three distinct routes:[7]
Langerhans cells’ conversion as they carry collected antigens to the lymph node, Myeloid precursor cell differentiation and Lymphoid precursor cell differentiation.
Histiocytic/dendritic cell sarcomas are exceedingly uncommon, and among those, IDCS is an uncommon type of unknown aetiology. Viral aetiology has not been supported, unlike FDC sarcoma (FDCS), where the EBV virus plays a vital role. Immunosuppressive drug use, particularly calcineurin, dysregulates the immune system, which reduces T-cell responsiveness to antigens presented by IDC.
Tumours pertaining to terminally differentiated B cells have been shown to undergo transdifferentiation into mature DCs by altering their transcription factor profiles. Therefore, this explains the fact that few cases of IDCS develop secondary to the transdifferentiation of B-cells, rather than the transformation of IDCs by themselves.[8] Individuals with IDCS during their lifetime can experience other haematopoietic and solid organ malignancies, as stated by Saygin et al.[9] There is no specific marker to diagnose IDCS because IDCs originate from multiple precursors. An overview of this tumour and adding it to the differentials of unusual-looking neoplasms are necessary to arrive at a correct diagnosis of IDCS. Differential diagnosis is quite challenging and includes both neoplastic and non-neoplastic entities. Nonetheless, the following differentials need to be taken into account: sarcomatoid carcinoma, melanoma, anaplastic large cell lymphoma, an IMT-like variant of FDCS, histiocytic sarcoma, Langerhans cell histiocytosis and inflammatory myofibroblastic tumours (IMT). IDCS does not reveal any distinct molecular abnormalities. A small number of solid and hematopoietic tumours have been found to harbour the BRAF V600E mutation. Rare examples of IDCS, Erdheim–Chester disease and Langerhans cell histiocytosis are among the histiocytic/dendritic cell tumours that encompass BRAF mutations. BRAF kinase inhibitor Vemurafenib is used in metastatic IDCS, according to Elisabetta et al. According to Xu et al., dendritic and histiocytic cell neoplasms express the programmed cell death 1 ligand (PD-L). IDCS is frequently passive to therapy and presents with extensive disease. Surgery is the primary treatment for localised diseases, and adjuvant therapies have no purpose. Chemotherapy is naturally used to treat metastatic illnesses. Several chemotherapeutic regimens of varying efficacy are used.[10] Poor prognostic factors include young age, co-occurring extranodal and nodal diseases, intraabdominal involvement and stage at the presentation, as highlighted by Saygin et al.[9]
Conclusion
Sarcomas of the histiocytic and dendritic cells are malignant tumours that have a high rate of morbidity and mortality. Additionally, IDCS is exceedingly uncommon and typically manifests at a late stage. For any unusual-looking neoplasm morphologically, certain diagnostic markers are to be used to establish and/or confirm the correct diagnosis.
Footnotes
Acknowledgements
We acknowledge the technical staff of the Department of Histopathology, Apollo Hospitals, Chennai.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Ethical approval
Approval was obtained from the Institutional Review Board (Ethics Committee) at Apollo Hospitals, Chennai, Tamil Nadu, India (Institutional Ethical Committee Approval Number AMH-C-S-056/07-23).
Informed Consent
Informed consent was obtained from the patient.
Credit author statement
KM: conceptualisation, data curation, formal analysis, writing the manuscript and critical revision of the manuscript.
MM: conceptualisation, supervision and critical revision of the manuscript.
Data availability
All data underlying the results are available as part of the article and no additional source of data required.
Use of artificial intelligence
Not applicable.