
Abstract

Dear editor, primary systemic vasculitides are well known for myriad manifestations due to inflammation of blood vessels. From the diagnostic and therapeutic perspective, primary systemic vasculitides are usually classified based on the size of the predominant type of vessels involved. Each type of vasculitis is known by some characteristic manifestations. It includes but is not limited to features like limb claudication or young hypertension in large vessel vasculitis, glomerulonephritis or prominent sinonasal symptoms in small vessel vasculitis and prominent neurological symptoms and gangrene in medium vessel vasculitis. However, there are several non-rheumatological conditions which can present with similar manifestations and thus can pose a diagnostic dilemma to the treating rheumatologist. Hereby, we present one such case which posed a diagnostic challenge to the treating team.
A 63-year-old lady, a known hypertensive, presented with a four-week history of progressive blackish discolouration and burning pain in the right little toe with swelling of the right lower limb. She had no history of fever, joint pain, weight loss or other systemic symptoms. She did not have any history of diabetes mellitus or any other systemic disease. She was taking amlodipine for her hypertension which was adequately controlled. Examination revealed stable vitals, blanchable purpura in the extremities and face, well-demarcated dry gangrene over the right little toe, and cutaneous infarct and ulceration over the dorsum of the left foot (Figure 1). All peripheral pulses were palpable. Neurological examination revealed normal power in all four limbs, and normal deep tendon reflexes but impaired proprioception over the distal part of bilateral lower limbs with other sensory modalities being unaffected. The rest of the systemic examinations were unremarkable.
(A) Digital Gangrene. (B) Cutaneous Infarct and Deep Ulceration Secondary to Rupture of Blebs on the Dorsum of the Foot. (C) Peripheral Blood Smear (400X) Showing Mild Leukothrmobocytosis. (D) Bone Marrow Biopsy (200X) Showing Cellular Marrow with Increased Atypical Megakaryocytes with Enlarged, Mature Hyper-Lobulated Nuclei.
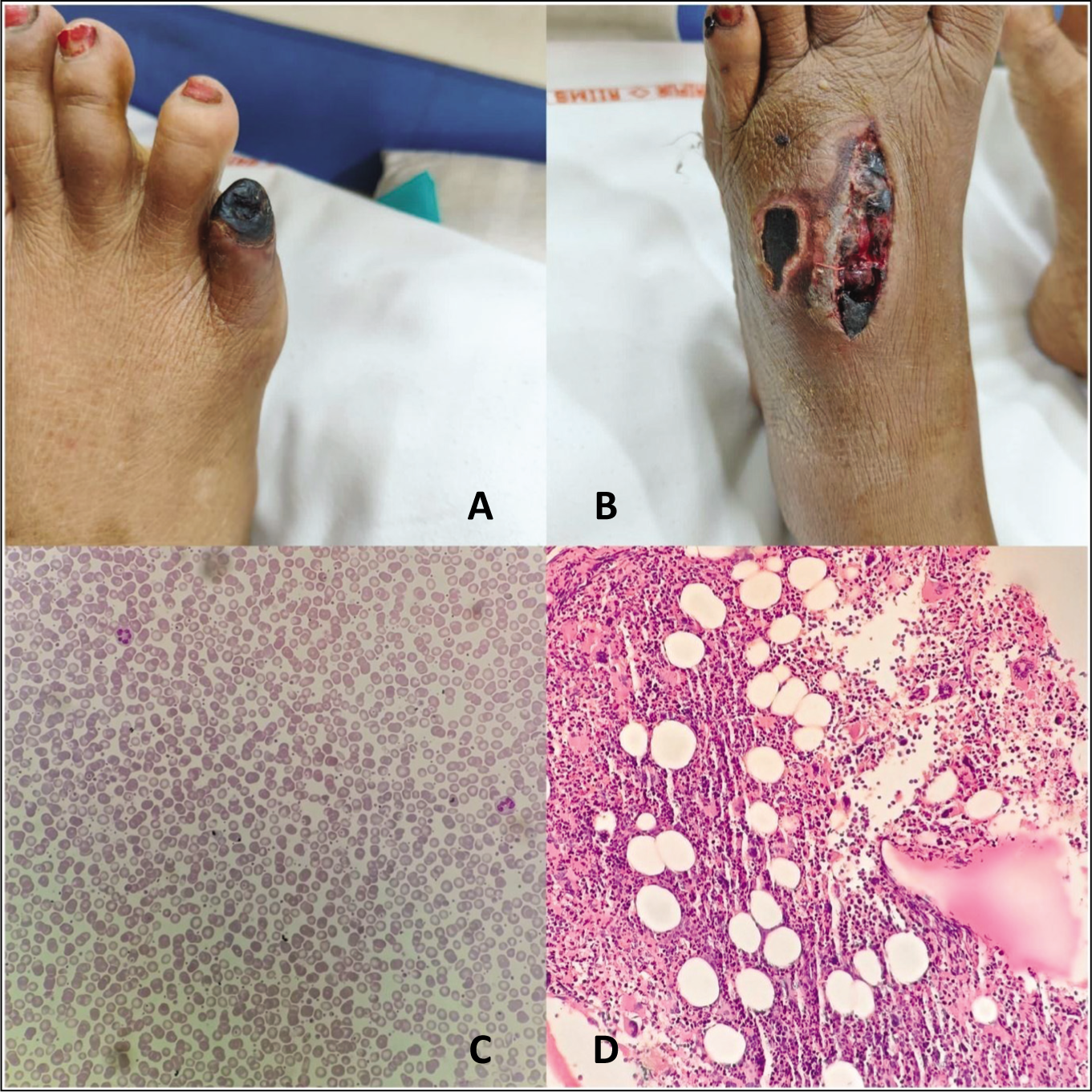
Considering the age of the patient, and the presence of comorbidity like hypertension, the possibility of peripheral arterial disease was considered. On the other hand, considering the cutaneous infarct, and abnormal neurological examination, primary systemic vasculitis like polyarteritis nodosa (PAN) and any thrombophilic condition like antiphospholipid antibody syndrome were considered as other possible differentials. Routine investigations revealed Hb of 7.8 gm/dL (Hct 53.8%), total leukocyte count (TLC) of 29,980/mm3 (N = 84%), platelet count of 5.89 lakh/mm3 and normal ESR and CRP, normal lipid profile and mildly deranged renal function (serum creatinine 1.54 mg/dL). Urine routine microscopy was unremarkable. All immunological workup (ANA, ANCA) were negative. The test for antiphospholipid antibody was negative. Arterial dopplers of both lower limbs and a CT angiogram of both lower limbs were normal.
Given normal arterial Doppler and normal lipid profile, peripheral arterial diseases seemed unlikely. Although high TLC with neutrophilic leukocytosis, elevated platelet count, mildly deranged renal function and abnormal neurological findings were more fitting with primary systemic vasculitis like PAN, a normal ESR/CRP was going against a diagnosis of PAN.
Diagnostic possibilities were reconsidered. Given persistently high levels of all three cell lineages in blood with high haematocrits, a possibility of polycythemia-induced gangrene was thought of. Bone marrow biopsy was done which showed markedly increased megakaryopoiesis (Figure 1). Further work-up revealed low levels of erythropoietin in serum and a positive JAK2V617F mutation. A diagnosis of polycythemia vera (PV) was made. Phlebotomy was done to bring down the red cell mass, and hydroxyurea was introduced, following which the rash subsided and the progression of gangrene halted once the haematocrit was brought down to below 42%. She is currently doing well on intermittent phlebotomy.
The presence of cutaneous and renal manifestations in the background of gangrene with soft neurological signs should heighten suspicion of primary systemic vasculitis especially medium vessel vasculitis. Vasculitides are a heterogeneous group of diseases that cause inflammation, primarily in the vessel wall with varying organ involvement depending on the type and size of the affected blood vessels. The first step in diagnosing a patient with vasculitis is to rule out other possible causes (mimics). 1 A thorough review of historical points, examination findings and laboratory parameters is essential to avoid misdiagnosis. Mimics of vasculitis maybe classified into those mimicking large, medium and small vessel vasculitides. 2 Medium vessel vasculitis typically affects the kidney, peripheral nerves and skin. 3 A large number of mimics of medium vessel vasculitis have been described. These include hereditary conditions like fibromuscular dysplasia, various hypercoagulable states, vasculopathies, cholesterol emboli, etc. 4 Although PV is a known hypercoagulable state, sometimes due to the subtleness of presentation, diagnosis can be challenging.
PV is a relatively indolent myeloid neoplasm that is typically suspected based on an incidental discovery in routine blood work which may have been done randomly. The most common symptoms which may be present at diagnosis include fatigue, pruritus, early satiety, concentration problems and inactivity. 5 It is the most common among the myeloproliferative neoplasms. PV typically has erythrocytosis, leukocytosis and thrombocytosis and results from a mutation in the JAK2 gene where valine is replaced with phenylalanine (V617F). This mutation (JAK2V617F) is expressed by more than 95% of patients with PV. Bone marrow examination shows hyperproliferation with increased erythroid, myeloid and megakaryocytic precursors. However, the bone marrow examination does not provide specific diagnostic information as it may be normal or indistinguishable from that of other myeloproliferative neoplasms like essential thrombocytosis and primary myelofibrosis. In cases where PV presents with elevated haemoglobin, haematocrit or red cell count alone, other causes of erythrocytosis should be considered. Although PV can cause haemorrhagic manifestations including major bleeding in 3 to 22% of patients, thrombotic manifestations are well reported which range from 12 to 25% in various studies.6,7 The thrombotic manifestations of PV are very important as PV may present with digital ischemia or cutaneous infarction similar to vasculitis and thus can mimic cutaneous predominant vasculitis. Moreover, leukocytosis and thrombocytosis can be seen both in PV and systemic vasculitis. A judicious clinical approach and careful analysis of laboratory findings like ESR, CRP and autoimmune serology can help in clinching diagnosis.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.