
Abstract
Introduction
Neu-Laxova Syndrome (NLS) is a rare, autosomal, and recessively inherited disease characterized by severe congenital malformations, leading to prenatal or early postnatal mortality. The hallmark clinical features of this syndrome are severe intrauterine growth restriction, central nervous system abnormalities, restrictive dermopathy, and characteristic facial dysmorphia. There is no cure or treatment for this syndrome and the termination of the pregnancy is the suggested approach.
Case description
Here, we report a new case of 2 siblings with NLS, diagnosed in utero by ultrasonography and genetic examination. Two variants in heterozygosity in the phosphoglycerate dehydrogenase gene were discovered, including a previously uncharacterized variant (c.487C>T p. [Arg163Trp]).
Comments
Despite all the advances in prenatal diagnosis, NLS remains undiagnosed in many pregnancies. Early and sequential US examinations should be performed in high-risk pregnant women, to establish an early diagnosis.
Background
Neu-Laxova Syndrome (NLS) is a rare, autosomal, and recessively inherited disease, characterized by severe congenital malformations that lead to prenatal or early postnatal mortality. 1 The hallmark clinical features of this syndrome are severe intrauterine growth restriction (IUGR), central nervous system (CNS) abnormalities, restrictive dermopathy, and characteristic facial dysmorphia. Additionally, limb malformations and generalized edema are frequently observed.2, 3 However, it is important to note that not all patients with NLS exhibit the same clinical features, which shows the great phenotypic heterogeneity that characterizes this syndrome. 4 Therefore, the diagnosis of NLS can be challenging and is often based on ultrasound (US) findings during prenatal screening. Confirmation of the diagnosis can then be done through genetic tests.5, 6 Unfortunately, there is currently no cure or treatment for NLS and due to its poor prognosis, termination of the pregnancy is often the suggested approach. 3 Here, we present a new case of NLS involving 2 siblings diagnosed in utero by US and genetic examination. These 2 siblings highlight the possibility of slightly different phenotypes associated with the same mutation.
Informed consent was obtained from the parents and ethical committee approval was taken for publication.
Case Report
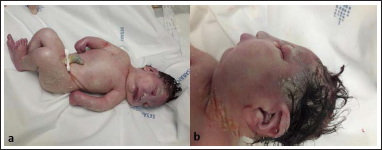
A 24-year-old pregnant woman, with a medical history of a total anomalous pulmonary venous return, heavy smoking, 2 previous spontaneous abortions (with another partner), and consanguinity to her current partner (first-degree cousin), was referred to a prenatal diagnosis consultation due to abnormal fetal ultrasonography findings at 13 weeks’ gestation. The US revealed a thicker neck translucency, retrognathia, and reverse return in the ductus venosus. Observations at 25 weeks’ gestation revealed a severe IUGR (0 percentile), microcephaly, edema, and flexion contractures of upper extremities. Cerebellar hypoplasia was also observed at 30 weeks. Fetal echocardiogram was normal. Subsequent prenatal biochemical screening revealed a low risk of trisomy and chromosomal analysis showed a normal karyotype (46, XY). The microarray was normal and the result for common aneuploidies was negative. Whole exome sequencing was performed and revealed a pathogenic variant (c.1468G>A p. [Val490Met]) and a variant of uncertain significance (c.487C>T p. [Arg163Trp]), both in heterozygosity, in the phosphoglycerate dehydrogenase (PHGDH) gene. The clinical phenotype showed extensive overlap with NLS and these variants were interpreted as the likely cause of the disease. The pathogenic variant (c.1468G>A p. [Val490Met]) was identified in the mother and the variant of uncertain significance (c.487C>T p. [Arg163Trp]) was identified in the father. The poor prognosis of the disease was discussed with the couple and they decided to maintain the pregnancy. At 41 weeks’ gestation, the woman gave birth to a male infant, with a birth weight of 1,585 g (0 percentile, -4.31 z-score, according to child growth standards of World Health Organization [WHO]) and an Apgar index of 3, 1, and 0 at the first, fifth, and tenth min, respectively. 7 No resuscitation measures were performed. On physical examination, we could see several features consistent with the NLS (Figure 1).
Two Photographs (A and B) of the First Baby with NLS Described in Our Case Report Showing Microcephaly, Micrognathia with Retrognathia, Flattened Nose with Broad Nasal Bridge, Full Cheeks, Large- and Low-set Malformed Ears, Short Neck, Swollen Hands and Feet, and Flexion Contractures of the Hands.
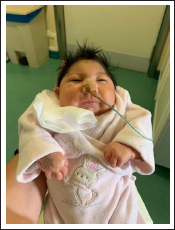
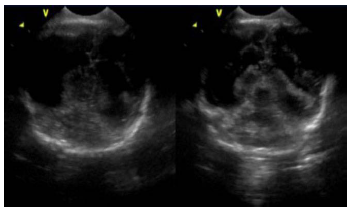
Five months later, the same woman, 12 weeks pregnant, returned for a follow-up visit. The US at 22 weeks’ gestation showed severe IUGR, micrognathia, and multiple CNS anomalies (ventriculomegaly, cerebellar hypoplasia, and partial agenesis of the corpus callosum). Fetal echocardiogram was normal. Genetic sequencing of this fetus revealed the same 2 variants found in the previous fetus. Other routine prenatal examinations were unremarkable. After the diagnosis of NLS, the parents decided to continue the pregnancy. At 39 weeks’ gestation, the woman delivered a female infant with a birth weight of 1,480 g (0 percentile, -4,53 z-score, according to child growth standards of WHO) and an Apgar index of 5, 9, and 10 at the first, fifth, and tenth min, respectively. 8 No resuscitation measures were performed. On physical examination, we observed a phenotype very similar to the previous baby (Figure 2), except for a complete cleft palate. Postnatal supportive and noninvasive care was provided. Postnatal transfontanellar US showed severe ventriculomegaly, agyria, and agenesis of the corpus callosum (Figure 3). There were no complications during hospitalization. Feeding was possible with a nasogastric tube, and the parents were educated on its use. Multidisciplinary care was provided including neonatology, gastroenterology, neurodevelopmental and physical medicine, and rehabilitation. At 37 days of age, the baby died at home.
A Photograph of the Second Baby with NLS Showing Severe Microcephaly, Micrognathia with Retrognathia, Full Cheeks, Low-set Ears, Short Neck, and Ichthyosis.
Postnatal Transfontanellar Ultrasonography Showing Severe Ventriculomegaly, Agyria, and Agenesis of Corpus Callosum.
It should be noted that prenatal counseling was provided during the first pregnancy and the risk of recurrence was explained to both the parents. Between both pregnancies, there was a loss of clinical follow-up due to poor parental adherence to medical consultations.
Discussion
NLS is a rare disease that was first named by Lazjuk et al in 1979 in an effort to unify the independent previous reports describing 3 affected siblings with several similar congenital anomalies.1 Each of these reports described 3 affected siblings with several similar congenital anomalies. 1 After describing 1 additional case, Lazjuk et al concluded that this might be a relatively rare and fatal autosomal recessive syndrome with malformations of 4 subgroups: incomplete differentiation, hypoplasias of several organs, deformities, and dysplasias. 1 At that time, the diagnosis was based on a clinical constellation of findings that were later confirmed at autopsy.
Today, we know that this syndrome has a wide range of complex clinical features that can be seen before and after birth. Antepartum abnormalities can aid in diagnosis of very early phase in pregnancy and may include polyhydramnios, reduced fetal movements, short umbilical cord, and small placenta in addition to severe IUGR.2, 3
Besides IUGR, the fetus/newborns may present with icthyosis, restrictive dermophathy (resulting in flexion contractures and immobilization of the major joints), microcephaly, and sloping forehead in 80% to 99% of cases during pregnancy and after birth. Other SNC anomalies include hypoplasia of the cerebellum (36%), agenesis of the corpus callosum (36%), and those described above (lissencephaly in 45% and ventriculomegaly in 17%); other dysmorphic features include hypertelorism (49%), flat or abnormal nose (79%), low or malformed ears, micrognathia (68%), and cleft lip or palate; ocular features such as proptosis (56%), ectropion, hypoplastic eyelids, microphthalmia, and cataracts; and deformities of digits and limbs such as rocker-bottom feet, among others.2, 3, 9 Genital underdevelopment, unilateral renal agenesis, pulmonary hypoplasia, and congenital heart defects may also occur but are less common. 2 There is some heterogeneity in the phenotype because not all of these signs and symptoms are present in every baby. As we could see in our case report, many of the characteristic findings of NLS were observed in both babies, but some different features were identified between the two.
Prenatal US is extremely useful for the diagnosis and subsequent management of NLS and is particularly important for monitoring high-risk pregnancies.10, 11 In our case, the first US, in the first pregnancy, was crucial for the timely diagnosis of this disease.
Similar cases have been reported previously. A history of parental consanguinity and recurrence in subsequent pregnancies is common. In fact, the majority of reported cases are from countries with high rates of consanguineous marriages, such as Morocco and Turkey.12–14 In our region, some cases of consanguinity between couples remain, as in the case presented here. In addition, the majority of the mothers had a history of miscarriage or stillbirth.10, 11, 13 In some cases, fetal death occurs in utero and in other cases, death occurs soon after birth. 4
NLS should be considered in the differential diagnosis of cases with IUGR, microcephaly, multiple dysmorphisms, severe joint contractures, restrictive dermopathy, ichthyosis, and nonimmune hydrops. The differential diagnosis includes trisomy 18 syndrome and severe arthrogryposis syndromes such as cerebroocular-facial skeletal syndrome, the lethal multiple pterygium syndrome, restrictive dermopathy, fetal akinesia/hypokinesia sequence, cérebroarthro-digital syndrome, harlequin fetus, Smith-Lemli-Opitz syndrome, and Miller-Dieker syndrome. 3
The recessive inheritance of this syndrome was confirmed in 2014 by the identification of homozygous mutations in PHGDH as the cause of NLS in 3 consanguineous families. PHGDH encodes for an enzyme involved in the first and limiting step of L-serine biosynthesis, resulting in L-serine deficiency. 14 High levels of this amino acid production are required for cell proliferation to replenish the one-carbon pool needed for the synthesis of nucleotides and other cellular components. The overall phenotype of these serine-deficiency disorders is determined by the level of residual enzyme activity, with NLS representing a more severe form.14, 15
In addition to mutations in PHGDH, NLS is known to be caused by mutations in phosphoserine aminotransferase 1 (PSAT1) and phosphoserine phosphatase (PSHS), both of which are involved in the same pathway of L-serine biosynthesis. NLS is currently classified as type 1 and type 2 when caused by mutations in the genes PHGDH and PSAT1, respectively. 13 Following these recent discoveries, some cases have already been described with the genetic study and the identification of the mutation involved.10, 16 Regarding genetic diagnosis, the initial chromosomal evaluation should be followed by the whole exome sequencing analysis, as verified in the case presented.
Currently, there is no cure or treatment for NLS. Some reports in the literature indicate that L-serine supplementation may prove to be an effective treatment in the future, if NLS is detected early in pregnancy or in subsequent pregnancies of parents who previously had a child with NLS. 13 For now, the approach to NLS after birth is symptomatic and supportive care, which may require the coordinated efforts of a multidisciplinary team, as was the case for the second baby described here. 3
Most cases of NLS have a poor prognosis. Many affected neonates are either stillborn, die soon after birth, or die in the first few weeks of life due to chest constriction, infection, or neurologic complications. 2 Given the poor prognosis of this condition, and the autosomal recessive inheritance, genetic counseling and characterization of the mutations are extremely important. 3 After prenatal diagnosis, the termination of the pregnancy is the suggested approach and the prevention can be achieved by in vitro fertilization and preimplantation genetic diagnosis. 12, 17
In the case described above, the same variants were found in both fetuses, and in both the parents. The first variant is a previously well-described pathogenic variant, and the second is a novel variant. Despite the uncertain significance of the second variant and in the absence of functional testing, both were considered as the likely cause of the presented phenotype.
Despite all the advances in prenatal diagnosis, NLS remains undiagnosed by many physicians. At least 90 cases have been described in the literature, many of them were diagnosed after birth. 9 Early and sequential US examinations are crucial and should be performed in order to establish an early diagnosis. It is important that potential new variants and new phenotypes are published in order to enrich the recognition of this disease.
Genetic counseling and psychological support for consanguineous carrier couples is crucial, and a comprehensive explanation of the disease prognosis is essential to limit the suffering of these families.
Footnotes
Author Contributions
Study design: Carolina Ferreira Gonçalves; data collection: Carolina Ferreira Gonçalves; data analysis: Carolina Ferreira Gonçalves; manuscript writing: Carolina Ferreira Gonçalves; manuscript revision: Alexandra Andrade, Patrícia Silva, Cremilda Barros, Carmo Camacho, and Edite Costa; study supervision: Alexandra Andrade, Patrícia Silva, Cremilda Barros, Carmo Camacho, and Edite Costa.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval from Institutional Ethical Committee was obtained.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
The participant has consented to the submission of the article to the journal.