
Abstract
This report is about a male newborn with pituitary stalk interruption syndrome (PSIS) who presented with apneic episodes associated with critical hypoglycemia at 3 hours of life (HOL). The onset of manifestations of PSIS varies in timing with the mean age of presentation between 4 and 5 years of age. The cause is unclear, but birth-related complications are considered potential factors. There is also a growing recognition of increasingly complex PSIS inheritance patterns. Further genetic workup for our patient revealed the deletion of the 16p11.2 gene with duplication of Xp22.33/Yp11.32. This is the first case report that describes PSIS in conjunction with these mutations, so the clinical correlation is unknown. However, 16p11.2 deletion is known to be associated with epilepsy and brain abnormalities. Additionally, duplication of Xp22.33/Yp11.32 is noted to be associated with short stature.
Keywords
Abbreviations
ASD: autism spectrum disorder
BG: blood glucose
CT: computed tomography
D10: dextrose 10%
FSH: follicle-stimulating hormone
GH: growth hormone
HOL: hours of life
IGF-1: insulin-like growth factor 1
IGFBP-3: insulin-like growth factor 1 binding protein-3
LH: luteinizing hormone
MRI: magnetic resonance imaging
PSIS: pituitary stalk interruption syndrome
SMA: spinal muscular atrophy
T4: thyroxine
TSH: thyroid stimulating hormone
Introduction
Pituitary stalk interruption syndrome (PSIS) is a congenital anomaly of the pituitary gland characterized by the triad of an interrupted (or a thinned) pituitary stalk, an ectopic (or absent) posterior pituitary gland, and hypoplasia/aplasia of the anterior pituitary gland. 1 The clinical manifestations of PSIS vary widely, including the age of onset (from newborn to adult) and the number of hormone deficiencies. According to reports, all PSIS cases are associated with a deficiency in growth hormone (GH), and almost 97.2% of cases have gonadotropin deficiency. 2 Corticotropin deficiency is reported in 88.3%, and thyrotropin deficiencies are reported in 70.3% of patients with PSIS. 2 Magnetic resonance imaging (MRI) is the preferred method for evaluating endocrine problems affecting the hypothalamic-pituitary region. 3
Case Presentation
A male baby was born at 41w0d via spontaneous vaginal delivery. Pregnancy was complicated by chlamydial infection at 25 weeks of gestation that was treated, followed by a test of cure. Maternal past medical history was significant for polycystic ovary syndrome, anxiety, and depression (on Bupropion). The delivery was complicated by the presence of a nuchal cord x 3. The baby received routine neonatal resuscitation per the neonatal resuscitation program and transitioned well with the mother.
At 3HOL the infant had an apneic event that required positive pressure ventilation support. Point of care (POC) glucose was 11 mg/dL with a subsequent serum glucose level of 9 mg/dL. The patient was given a D10 bolus and then started on a D10 infusion, given the persistent hypoglycemia. The POC glucose improved to 96 mg/dL. Oral glucose and formula supplementation were provided. He had 2 subsequent apneic episodes at 7HOL and 18HOL, both of which were associated with low BG readings. Each time the patient received an additional intravenous bolus of D10, the rate of D10 infusion was increased accordingly. CT brain without contrast was performed, which showed an intracranial subdural hematoma along the tentorium and within the posterior fossa with no focal parenchymal hemorrhage, parenchymal mass effect, or edema. Full septic work up (CBC, CRP, blood culture, HSV PCR, and lumbar puncture) was performed to rule out infectious cause of hypoglycemia. The infant was transferred to our hospital at ~34HOL for further management.
Upon arrival, the patient was noted to have significant jaundice. Otherwise, the physical exam was unremarkable, including descended testes bilaterally, normal size of the phallus, and no syndromic facial features. The baby received a total of 48 hr of empiric ampicillin, gentamycin, and acyclovir which were discontinued after the septic work up came back negative. Given the persistent hypoglycemia despite formula fortification and even continuous feeds at 24 kcal/oz, further workup was initiated on DOL 6. Critical labs were drawn in the setting of BG level of 37–47 mg/dL, and they were significant for low cortisol (1.3 mcg/dl) and low GH (1.09 ng/ml). ACTH stimulation test was performed and consistent with adrenal insufficiency. A glucagon stimulation test was not consistent with an insulin-mediated process (initial BG level 32 mg/dL, glucagon 0.1 mg was given followed by a rise of BG by ~ 10 mg/dL). Ammonia, free fatty acids, carnitine were reassuring, making inborn errors of metabolism less likely. MRI of the brain with pituitary cuts showed an ectopic posterior pituitary gland at the level of the hypothalamus; a definitive infundibula/pituitary stalk was not entirely visualized. The baby was diagnosed with PSIS and was started on a maintenance dose of hydrocortisone with subsequent improvement of hypoglycemia. He also was started on levothyroxine on DOL21 after the thyroid studies resulted (low TSH and free T4). Both initial and repeat newborn screens came back normal.
During his follow-up visits, he had poor weight gain (<3rd percentile) with a subsequent decrease in growth velocity; despite being on 24 kcal/oz formula. At 4 months of age, GH surrogates (IGF-1, IGFBP-3) were performed and showed low IGF-1 <10 ng/mL, IGFBP-3 0.9 ug/mL, and thus was started on GH supplementation. A genetic evaluation (single nucleotide polymorphism [SNP] chromosomal microarray, Russell-Silver syndrome analysis) demonstrated deletion of 16p11.2, consistent with a diagnosis of 16p11.2 deletion syndrome. Additionally, his SNP chromosomal microarray identified a duplication of Xp22.33/Yp11.32. He was anticipated to be a spinal muscular atrophy carrier based on the deletion of SMN1.
Discussion
PSIS is a rare condition with an incidence of 0.5/1,000,000 births. 4 The onset of manifestations varies in timing. Clinical symptoms often appear in the first decade of life, but diagnosis is frequently delayed. 5 In our case, the patient presented as early as 3HOL and was diagnosed at DOL 6. There were just 3 other cases with such an early diagnosis and relatively similar severity of presentation described in the literature in the case series written by Mehta et al. 6 Most other cases were diagnosed later in life as the symptoms were less pronounced. Deficits may evolve with the onset of new pituitary hormone deficiencies that progress to panhypopituitarism. 5 Our patient’s initial symptoms resolved after treatment with hydrocortisone, then he was started on Levothyroxine and later in life required GH supplementation. It is possible that when he reaches a pubertal bone age, he will also require testosterone supplementation.
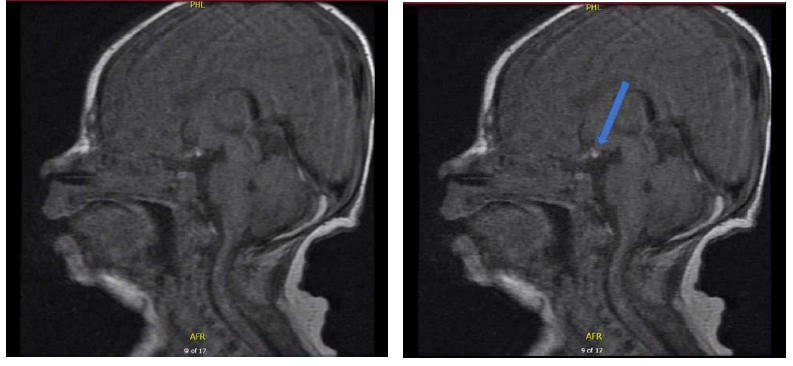
Pituitary MRI is the modality of choice for diagnosis since it allows detailed and precise visualization with differentiation of anterior and posterior pituitary lobes. A marker of neurohypophysial functional integrity is the identification of posterior pituitary hyperintensity in the posterior part of the sella turcica. 3 Image 1 represents the MRI results for our patient.
Ectopic Posterior Pituitary Gland. A Definitive Infundibula/Pituitary Stalk Is Not Entirely Identified.
There is growing recognition of genetic mutations in some cases of PSIS (mutations in HESX1, LHX4, PROP1, OTX2, SOX3, PROKR2, GPR161, CDON, ROBO1 genes),1, 7 which prompted further genetic workup for our patient.
16p11.2 deletion is one of the most common genetic causes of autism spectrum disorder and other neurodevelopmental disorders with an incidence of approximately 1/2000. 8 It is also associated with morbid obesity, macrocephaly, and epilepsy. 9 Congenital abnormalities are also more common in children with this syndrome. 10 A study performed on mice by Ouellette et al, suggested that 16p11.2 deletion leads to male-specific, endothelium-dependent structural and functional neurovascular abnormalities. 11
Chromosomes Xp22.33 and Yp11.32 contain a homeobox-containing (SHOX) gene. More than 300 different mutations in the SHOX gene responsible for short stature have been described and considered one of the most common genetic causes of short stature. 12
This is the first case report that describes PSIS in conjunction with 16p11.2 deletion syndrome and duplication of Xp22.33/Yp11.32. It is unclear if the co-existence of those mutations contributed to such an early and severe presentation of PSIS in this patient. However, it can serve as a starting point for future research with the primary interest in correlating PSIS and the described mutations; the latter of which have known associations with brain abnormalities and short stature.
Footnotes
Author Contributions
All authors contributed to conception and design of the manuscript, analysis, and interpretation of data, drafting the article, reviewing, and revision of the manuscript.
Ekaterina Iausheva—drafting the article and further editing it.
Sara Mohamed—drafting the article and further editing it.
Elizabeth E. Littlejohn—revising the article and final approval of the version to be published.
Tarek Mohamed—revising the article and final approval of the version to be published.
Mohammed Abdulmageed—revising the article.
All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
As this manuscript is a case report, no human or animal studies were performed.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
Written informed consent was obtained from the parent to publish the details of the child’s medical case and any accompanying images.