
Abstract
Background
Autoimmune glial fibrillary acidic protein (GFAP) astrocytopathy is an immune-mediated inflammatory disease of the central nervous system, the clinical phenotype of which includes meningoencephalitis, myelitis, optic neuritis, seizures, movement disorders, autonomic dysfunction among others, affecting people across all ages. Due to its recent discovery, there is a paucity of literature on this topic and an absolute lack of Indian case series.
Purpose
This study aims to shed light on the variable presentations of anti-GFAP astrocytopathy and review the existing literature on the topic.
Methods
This was a retrospective study that included all patients who tested positive in the cerebrospinal fluid(CSF) and/or serum for GFAP immunoglobulin G between February 2023 and August 2023, after obtaining ethical clearance. Relevant clinical, demographic data was collected from the electronic medical records. A descriptive analysis of data was done and the current available literature was reviewed.
Results
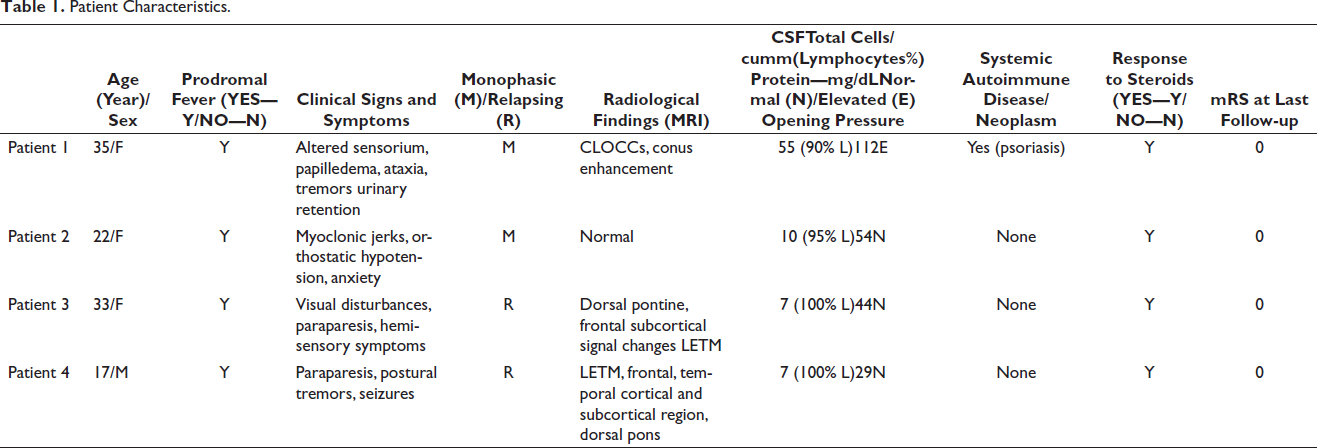
Our case series included four patients (F:M::3:1) with a median age of 28 years at symptom onset. Two of the cases had a relapsing-remitting disease pattern, while the other two had monophasic illnesses. The clinical spectrum we encountered included ataxia, tremors, myoclonus, seizures, recurrent myelitis, brain stem syndromes, autonomic dysfunction and psychiatric manifestations. All four patients responded remarkably to steroids and two patients are on rituximab therapy.
Conclusion
Autoimmune GFAP astrocytopathy encompasses an expanding clinical spectrum and should be considered in the context of myelitis, optic neuritis, ataxia, papillitis, seizures, autonomic dysfunction and movement disorders occurring in isolation or more commonly in varying combinations. Our case series, the first in India, shows a favourable clinical profile and the primary hurdle encountered in all four cases was to establish a diagnosis, further stressing the need for a predictive diagnostic algorithm.
Introduction
Autoimmune glial fibrillary acidic protein (GFAP) astrocytopathy is an immune-mediated inflammatory disease of the central nervous system originally described in 2016. 1 Since the discovery of the antibody responsible for the disease process, the clinical phenotype has expanded to include myelitis and optic neuritis in addition to meningoencephalitis affecting people across all ages and ethnicities.2–7 Due to its recent discovery, there is a paucity of literature on this topic and an absolute lack of Indian case series. This study aims to shed light on the variable presentations of anti-GFAP astrocytopathy through the descriptive analysis of four antibody-positive cases and review the existing literature on the topic.
Methods
This was a retrospective study that included all patients who tested positive in the cerebrospinal fluid (CSF) and/or serum for GFAP immunoglobulin G (IgG) between February 2023 and August 2023 who were then followed up and retrospective data collected. Ethical clearance was obtained from the institutional ethics committee. Relevant clinical and demographic data was collected from the electronic medical records of the institute. Neurological examination was performed by two neurologists independently. Imaging data was reviewed by the radiology team of the primary institute and by a radiologist from a different institute. Serological and CSF testing for the anti-GFAP antibodies was done at the Neuroimmunology laboratory, Amrita Institute of Medical Sciences, Kochi, using tissue-based assay by indirect immunofluorescence technique. Additionally, the patients also underwent testing for Aquaporin 4 antibodies, myelin oligodendrocyte glycoprotein (MOG) antibodies, autoimmune encephalitis panel, anti-nuclear antibody (ANA) profile and screening for underlying malignancy.
During the study period, our institute sent seven samples for GFAP testing among which four tested positive (details in Supplementary File).
We then performed a descriptive analysis of our data using percentage, mean and median. To review the current available literature, we searched the PUBMED database with the keywords autoimmune GFAP astrocytopathy and included original case series from various countries in addition to relevant case reports obtaining an extensive overview of the disease.
Results
Case 1
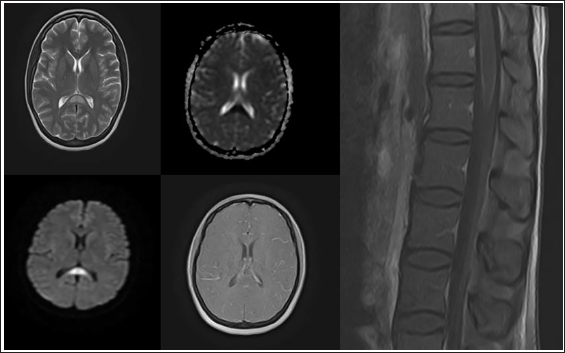
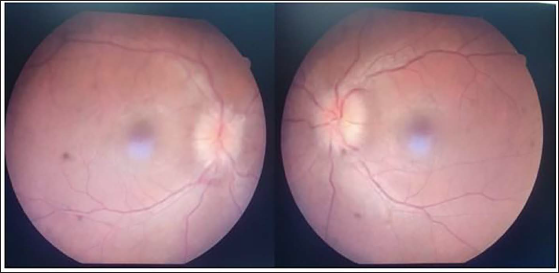
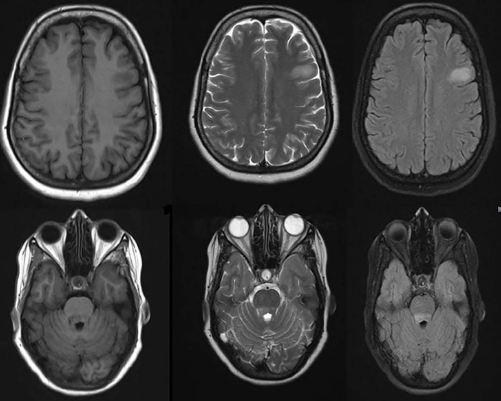
A 35-year-old lady, with a history of psoriasis, presented to an outside hospital with a history of fever and headache of two days duration followed by altered sensorium, was taken to the emergency room and was found to have severe hyponatremia (115 meq/L). She was initiated on antipyretics for fever and her sodium was corrected. She was also found to have a retention of urine and was catheterised. MRI brain with contrast including the spine showed a cytotoxic lesion of the corpus callosum (CLOCC) with leptomeningeal enhancement of conus (Figure 1). LP CSF was done which showed lymphocytic pleocytosis and elevated protein levels (cell count 135—88% lymphocytes, protein 82 mg/dL and glucose 49 mg/dL). After recovery of the sensorium, she was noticed to have persistent urinary retention, diplopia and gait ataxia. She was extensively evaluated for the above symptoms and all tests including autoimmune encephalitis workup, Aquaporin 4 and MOG antibodies, viral encephalitis panel and toxic screen (blood and urine) were negative. She was managed with empirical antibiotics and discharged. Post discharge patient had persistent diplopia, mild ataxia and urinary retention for which she would void via intermittent self-catheterisation. She presented to our hospital in the fourth week of her illness for further evaluation. On our clinical assessment, she was noted to have bilateral papilledema (Figure 2), impaired tandem gait, postural tremors and urinary retention. MR Imaging of the brain and spine was repeated which showed a resolving splenial lesion but had persistent leptomeningeal enhancement of the conus (Figure 3). CSF was repeated which showed elevated opening pressure of 340 mm of CSF, reducing trend of lymphocytic pleocytosis and persistently elevated proteins with normal sugar (56 cells, 90% lymphocytes, protein 112 mg/dL, glucose 60 mg/dL). Repeat serum Aquaporin 4 and MOG antibodies were negative. At this point, the common causes for inflammatory and infectious encephalomyelitis were ruled out (details in Supplementary File) and the possibility of autoimmune GFAP astrocytopathy was considered. Serum and CSF GFAP antibodies tested via indirect immunofluorescence were strongly positive. The patient was initiated on pulse methylprednisolone (1 g/day) for a total duration of three days followed by a tapering dose of oral steroids. She made a gradual and complete resolution of diplopia, disc oedema, gait symptoms and urinary retention by the fifth week of initiating steroids.
MRI Brain Showing T2 FLAIR Hyperintensity with Diffusion Restriction and No Contrast Enhancement in the Splenium of Corpus Callosum. MRI Spine Showing Leptomeningeal Enhancement of Conus.
Fundus Photograph Showing Bilateral Papilledema.
Serial MRI Imaging Done After Three Weeks Showing Resolving Splenial Lesion and Persistent Leptomeningeal Enhancement of the Conus.
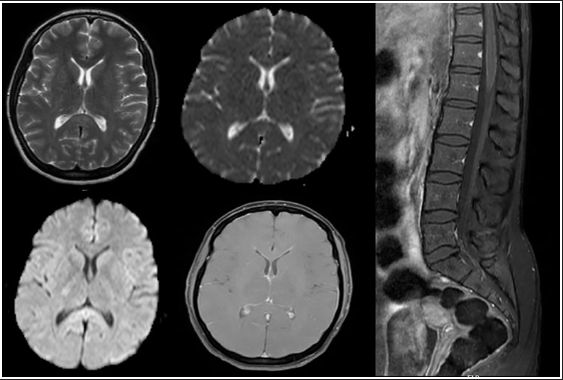
At the last follow-up, eight weeks post-symptom onset, the patient had remained asymptomatic and was not on any medications.
Case 2
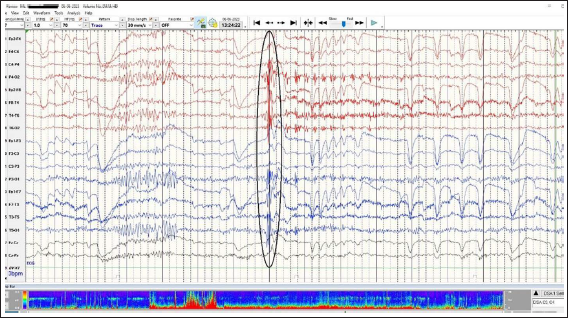
A 23-year-old lady presented to us with h/o imbalance while walking and occasional myoclonic jerks. There was a history of mild COVID three weeks before symptom onset for which she received treatment on an outpatient basis. She also reported being anxious and agreed to have some stressors. On examination, her neurological examination was unremarkable except for difficulty while performing tandem gait. During the clinical examination, frequent myoclonic jerks were observed. As she had mild ataxia only on attempted tandem gait in isolation, anxiety, sleep disturbances and stressors psychiatrist’s opinion was sought for the same. Prior to this, she had been evaluated at an outside centre, during which she was noted to have paroxysmal jerky movement of the eyeball (subjective), and opined to have doubtful psychogenic ataxia with vertigo. On one occasion she had severe postural giddiness and was documented to have orthostatic hypotension (recorded SBP 80 mm Hg). She made a spontaneous recovery from orthostatic giddiness over the next few days. When she came to our centre, the above-mentioned symptoms had resolved except for the mild ataxia and myoclonic jerks. For the evaluation of myoclonic jerks, a Video EEG was done, which showed a generalised discharge during a myoclonic jerk (Figure 4). She was then evaluated for possible autoimmune aetiology (as all symptoms started following COVID), Serum and CSF GFAP antibodies and serum anti-IgLON5 antibodies were sent. She was found to be positive for GFAP antibody (both serum and CSF). The patient was prescribed oral steroids with which her symptoms completely resolved over the next three weeks. The last follow-up was in the sixth week, she has been asymptomatic and not on any medications.
EEG Showing Generalised Spike Wave Discharge. Eye Blink Artifacts were also Noted.
Case 3
A 37-year-old lady who was being managed at an outside hospital for seronegative neuromyelitis optica (Aquaporin 4 and MOG antibody negative), symptomatic since 2019 was on follow-up in our institute from October 2020. Her first clinical presentation was left optic neuritis followed by longitudinally extensive thoracic transverse myelitis (LETM) in November 2019. Before presenting to us, the patient had received two doses of rituximab (December 2019, August 2020), the second dose of which was delayed by two months and preceded by a clinical relapse in the form of paraparesis. On our follow-up patient was administered a maintenance dose of rituximab (March 2021, September 2021) and she remained in remission till she was temporarily lost to follow-up, missing her subsequent dose. She then presented to us in July 2023 with right-sided sensory paresthesias and right lower limb weakness preceded by a self-limiting febrile episode. CSF done showed seven cells, 100% lymphocytes and a protein of 44 mg/dL. MRI brain revealed T2/FLAIR hyperintensity in the left frontal subcortical white matter and dorsal pons (Figure 5). A repeat serum Aquaporin 4 and MOG antibodies along with GFAP antibody testing was done. MOG antibodies, which had been negative twice before and the GFAP antibodies tested positive. The patient made a complete clinical recovery following a pulse of methylprednisolone and was then restarted on a maintenance dose of rituximab.
MRI Showing T2/FLAIR Hyperintensity in Left Frontal Subcortical White Matter, Grey White Matter Junction and Dorsal Pons.
Case 4
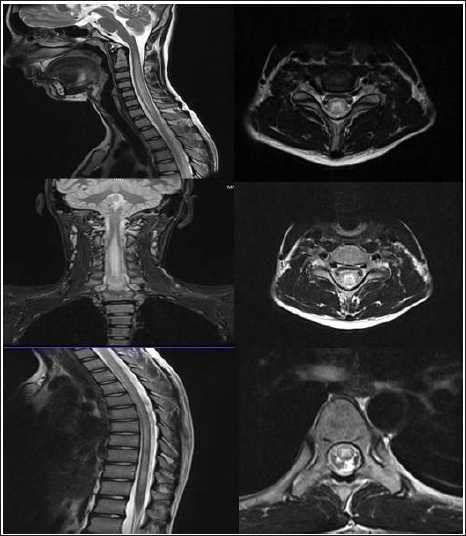
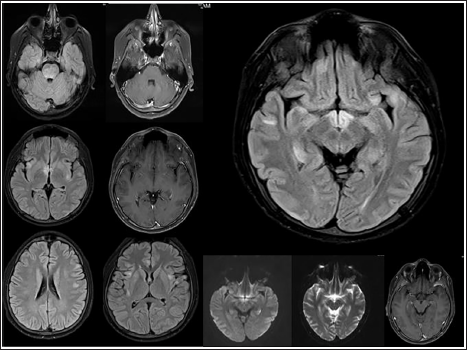
A 19-year-old boy presented with a history of relapsing-remitting neurological illness since Nov 2021. At symptom onset in November 2021, he presented with a history of lower limb weakness, sensory loss below the chest and bladder disturbance of two days duration. Acute transverse myelitis was suspected and MRI was done which showed longitudinally extensive hyperintensity extending from C2 to D11. Serum NMO-MOG antibody and ANA profile were negative. Lumbar puncture was not performed as consent could not be obtained. He was treated with an injection of methylprednisolone 1 g/day for five days followed by oral steroids. He showed significant improvement by the third week of steroid initiation. Steroids were continued for 10 weeks and gradually tapered. However while on tapering doses, at a dose of 10mg prednisolone per day, the patient had a clinical relapse in the form of recurrence of lower limb weakness and bladder disturbance. CSF during this time showed a cell count of 7 (100% lymphocytes) with normal protein levels (29 mg/dL) The steroid dose was optimised again and was initiated on a steroid-sparing agent mycophenolate mofetil (MMF) 500 mg/day. The dose of mycophenolate was gradually optimised to 1,500 mg/day, despite which in July 2022 the patient had a third relapse, with MRI showing longitudinally extensive cord hyperintensities from C3–D3, D6–D7, D10–D11 (Figure 6). He was given pulse steroids for three days and initiated an induction dose of rituximab. In December 2022 and January 2023, the patient had episodes of lower limb weakness (each lasting for one week) and required pulsing with methylprednisolone. In February 2023 patient went to another centre where he was reinitiated on mycophenolate and rituximab discontinued. The patient remained asymptomatic till September 2023 when he presented with an episode of generalised tonic-clonic seizure followed by confusion and anterograde memory loss lasting for about 72 hours. It was preceded by a febrile illness one week back. MRI done showed new T2/FLAIR hyperintensities in ventral pons, thalamic-mesencephalic junction and cortical and sub-cortical areas (Figure 7). The clinical phenotype of recurrent steroid-responsive myelitis in the absence of aquaporin or MOG antibodies prompted us to investigate for possible underlying GFAP astrocytopathy and serum antibodies tested were strongly positive. The patient was administered pulse steroids for three days along with anti-seizure medications, following which he made a complete recovery. He was then restarted on a maintenance dose of rituximab.
MRI Spine Showing Extensive Cord Signal Changes in the Form of T2 Hyperintensity Extending From Cervical (C2) to Thoracic (T11) Segments with Involvement of Grey Matter (H pattern) Noted in Axial Sections.
T2/FLAIR Hyperintensities in Ventral Pons, Thalamo-mesencephalic Junction, Frontal and Temporal Cortical, Sub-cortical Regions.
Discussion
GFAP is an intermediate filament protein expressed in astrocytes that regulates its shape and motility and is involved in synaptic plasticity and reactive gliosis. 8
The antibody to this protein was first isolated by researchers at the Mayo Clinic in 2016 1 when analysing the samples of previously uncharacterised seronegative autoimmune encephalitis and other neuroinflammatory conditions over 18 years.
The median age of symptom onset in this study was 42 years with two subsequent case series having a median age of 44 and 43 years, respectively.2, 3 Studies in China, 4 Italy 5 and Japan 6 have shown a median age of 54, 52 and 44 years, respectively.
Our case series has a relatively younger median age of onset of 28.5 years (range 17–33). While most of the case studies so far do not show any gender predilection, we had a female preponderance (3/4, 75%). The demographic variability in our study is likely due to the paucity of the number of detected cases in India.
Our understanding of the clinical phenotype has widely expanded since the primary study was published. The most common clinical spectrum encountered in the original study was meningoencephalitis with or without myelitis, seen in all the initial 18 cases. 1 Before the isolation of this antibody, these cases had variably been classified under the clinical entity ‘Nonvasculitic autoimmune inflammatory meningoencephalitis’. 9 Headache of a subacute onset was a prominent symptom that was seen in only one of our patients. The patients were also noted to have papillitis without evidence of raised intracranial pressure. This finding was further confirmed in other case series as well. The reason behind the papillitis is likely due to the expression of GFAP in the retina by astrocytes and associated inflammation. 10
Distinct from the originally described optic disc oedema without raised intracranial pressure, the first patient of the current series had documented papilledema with raised intracranial pressure. Other case series have noted evidence of raised intracranial pressure2, 6 as well.
While none of the originally described cases had seizures, two patients among the Chinese cohort had seizures and additional studies have revealed that seizures are a prominent clinical presentation.3–6 The second patient in our cohort had myoclonic jerks, while the fourth patient had generalised seizures.
Movement disorders were a common clinical presentation in the studies of China, Italy and Japan occurring at a rate of 15.8%, 13.6% and 50%, respectively,3–5 with the most common ones being dyskinesias, tremors, choreoathetosis and myoclonus. Kimura et al. specifically studied movement disorders in GFAP astrocytopathy and his analysis revealed that among the patients 85% (N 74) had movement disorders including ataxia, tremor, myoclonus, dyskinesia, opsoclonus, rigidity, myokymia and choreoathetosis in decreasing order of occurrence. 11 Two of our patients had postural tremors and two had ataxia at symptom onset.
In summation, the clinical spectrum documented among our cases included meningoencephalitis, ataxia, tremors, autonomic dysfunction in the form of urinary retention and orthostatic hypotension, recurrent myelitis and papilledema with evidence of raised intracranial pressure (Table 1). Additionally, the second patient had episodes of anxiety and lack of sleep. Psychiatric manifestations occurring in this disease have been best described by the case reported by Zarkali et al., where irritability and aggression were the major symptoms at presentation. 7 Rarer clinical manifestations of this disease include motor-predominant peripheral neuropathy,12, 13 progressive cognitive impairment 13 and area postrema syndrome.14, 15 In all our cases, more common causes of the above clinical spectrum like vasculitis and sarcoidosis were ruled out (details in Supplementary File).
Patient Characteristics.
The characteristic radiological finding most commonly encountered in GFAP antibody-associated astrocytopathy is a linear perivascular enhancement oriented radially to the ventricles. Case series by Flanagan et al., Long et al., Kimura et al. and Dubey et al. showed this finding in 53%, 2 42.1%, 4 28% 6 and 51% 16 of their total cases, respectively. This finding is included as a radiological red flag sign when evaluating a patient suspected to have neuromyelitis optica spectrum disorder (NMOSD). 17 Other radiological findings include longitudinally extensive transverse myelitis affecting the central cord,5, 6 diffuse leptomeningeal enhancement, 7 thalamic and brain stem hyperintensities. 16 The myelitis noted among our patients did not have significant cord swelling or necrosis on the MRI in comparison to NMOSD cases.
Additionally, among patients with isolated ataxia, cerebellar atrophy has been observed.3, 4
Our patients did not have the pathognomonic MRI finding, however, we observed three distinct imaging features. The first patient at initial symptom onset had a CLOCC which resolved in the subsequent MRI with persisting enhancement of the conus. While the latter is a commonly documented finding in GFAP astrocytopathy, CLOCCs in this clinical setting is a relatively rarer radiological finding. 18 Among the French cohort, there were four cases with similar MRI findings of CLOCC which resolved completely in two of the cases and partially in the other two, requiring a median period of two to six months for the same. 3 CLOCCs have been previously described in several neuro infections, 19 further supporting the hypothesis of infections acting as a trigger for GFAP astrocytopathy.16, 20
Our second case has normal radiological findings which is seen in around 18% of such cases. 2 This case is unique in itself as the patient has subtle clinical signs in the form of myoclonus against a background of transient dysautonomia, psychiatric symptoms and a normal MRI.
The third patient, having a relapsing disease, over the years had radiological features of optic neuritis, myelitis and most recently frontal subcortical and dorsal pontine signal changes. Radiologically her MRI findings were distinct in that the most recent relapse displayed a frontal subcortical involvement. Another unique aspect of this case was that serum antibodies to MOG were also positive. There have been documented cases of coexisting AQP4 IgG cases in the clinical context of LETM (2/6 of LETM2). Among the French cohort, three patients had associated coexisting MOG-IgG or AQP4-IgG antibody positivity. 3 Additionally, an association of anti-MOG Ab has also been described in one Chinese patient with coexisting AQP4-IgG. 21 The coexistence of these antibodies indicates a unifying mechanism behind these neuroinflammatory conditions and whether there are prognostic implications of the same remains elusive.
The fourth patient had features of LETM in the serial MRIs performed during each relapse. During the latest relapse, signal changes were noted in the pons, thalamo-mesencephalic junction and cortical areas. The involvement of these regions has not been reported earlier in this disease.
Routine CSF findings in most of the initial Mayo cases (13 of the 14 available) showed an inflammatory picture with lymphocytic pleocytosis and marginally elevated proteins with five patients showing supernumerary oligoclonal bands. 1 Case series by Flanagan et al. and Long et al. showed similar abnormal CSF findings in 88%, 2 100% 4 among their patients. A unique finding noted in the study by Kimura et al was the presence of elevated adenosine deaminase levels in the CSF. 6 In our series, the CSF picture of the first patient resembled the classic pattern of lymphocytic pleocytosis with elevated protein levels, while the second and fourth ones showed mild lymphocytic pleocytosis with normal proteins.
In the third case, there were seven lymphocytes detected with normal proteins, however, oligoclonal bands were also detected.
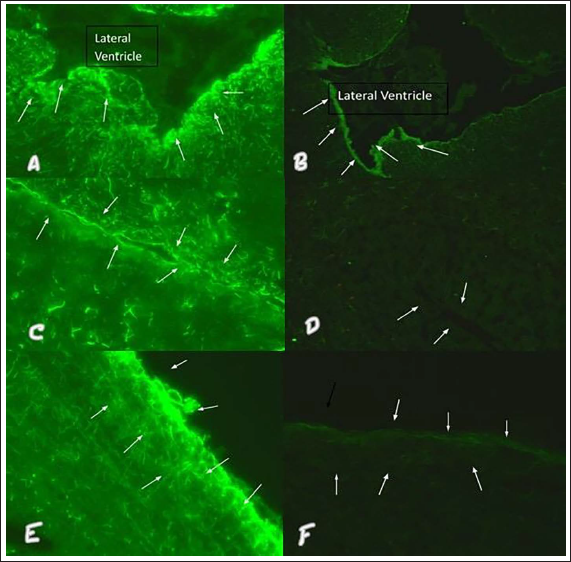
Serological confirmation in most of the major case series was done both by cell and tissue-based techniques.1–3,5 While the original study demonstrated predominantly pial, subpial and periventricular antibody binding 1 the Chinese study demonstrated an IgG immunostaining pattern in the molecular layers with a Bergmann radial pattern. 4 Among our patients, we demonstrated both patterns of binding (Figure 8).
Brain Parenchyma Adjacent to Lateral Ventricle (Arrows) Showing Strong Positive (Green Fluorescence) with GFAP IgG Using Rat brain Frozen Section in (A) Serum Sample 1: 10 Dilution—Magnification 40×. (B) CSF Sample Undiluted—Magnification 20×. Bergmann Radial Pattern in Strong Positive GFAP IgG CSF in (C) Serum Sample 1: 10 Dilution—Magnification 40×. (D) CSF Sample Undiluted—Magnification 20×. Pia and Subpial Region of Cerebral Cortex Showing Strong Positive (Green Fluorescence—Arrow) GFAP IgG in (E) Serum Sample 1: 10 Dilution—Magnification 40×. (F) CSF Sample Undiluted—Magnification 40×.
Since its description in 2016, there has been an added focus on attempting to identify the specific trigger leading to antibody production. Among the Mayo Clinic patients 29% of patients had preceding flu-like symptoms 1 while other studies have shown a range of 40%–60%. 22
In our case series, both symptom onset and relapses were preceded by fever, one of the infections being COVID. To our knowledge, this is only the second reported case of COVID-19 triggering GFAP astrocytopathy. 23 In 2021 Cheng et al. reported a case report of concomitant COVID and adenovirus triggering GFAP astrocytopathy 24 and Koh et al. reported an isolated case following COVID vaccination. 25 Isolated case reports of Epstein–Barr virus 26 and neuroborreliosis 27 infections preceding this astrocytopathy have also been reported.
There is still unclear data to suggest whether the majority of the cases are relapsing or monophasic. While the original study gave a relapse rate of 45% 1 , the French cohort had a favourable clinical picture with 82.5% of patients having only a monophasic illness. In the latter case, the relapses occurred after or during steroid taper similar to our two cases. 3 It must be noted however that 2/3rd of those patients were on long-term steroid-sparing agents.
Two of our cases were monophasic while the other two were relapsing remitting. In both the patients with relapsing clinical illness, each relapse was preceded by an infection and responded favourably to steroids.
Most of the case series, similar to ours, have shown a favourable and robust response to steroids.1–3,6 The mechanism for this exquisite steroid responsiveness is postulated to be secondary to the susceptibility of dendritic cells (DC). The DC which are potent antigen-presenting cells are highly sensitive to corticosteroids and abundantly located in the meninges, the perivascular spaces, and the juxta vascular parenchyma, that is, all the sites preferentially affected in GFAP astrocytopathy. 28 Another proposed explanation for this phenomenon is that GFAP-specific IgG may be accompanied by another antibody targeting the plasma membrane of the astrocyte. 5
Overall relapse rates are quite low. The patients in Japan did not have any relapse in the follow-up period and the only residual symptom was urinary retention in one patient while the prospective study in the US showed a relapse rate of only 18%. 12 The 19 patients in the Chinese study, however, had a sub-optimal response to both steroids and IVIg and a worse disease severity was noted, with some patients developing a progressive disease. 4 Ethnicity-dependent susceptibility factors could play a role and further study into this is warranted. Among the cases that were prospectively evaluated by Dubey et al., concurrent NMDA-R-IgG positivity and underlying cancer were associated with a lack of response to first-line Immunotherapy. 12
All the major studies show a varying percentage of patients with a systemic autoimmune disease or underlying neoplasm. Seven of the original 16 patients had other autoimmune conditions including endocrinopathies, myositis and interstitial pneumonitis. 1 The study in China showed a much higher incidence of coexisting autoantibodies of 76.4% 4 while in Italy it was just 27% with three patients having rheumatoid arthritis. Six patients had an underlying neoplasm, ovarian teratoma and prostate adenocarcinoma. Other case series have revealed a 22%, 2 14% 5 association of a coexistent or subsequently discovered neoplasm. Additionally, simultaneous detection of anti- NMDA receptor and Aquaporin 4 antibodies made an underlying teratoma more likely. 16
Only one of our patients had psoriasis (well controlled) while other coexisting autoimmune disorders or an underlying neoplasm were not uncovered in any of our patients and all four were screened for the same.
The most commonly encountered systemic complication seen in the previous case series1, 16 was hyponatremia seen in one of our patients. A unique finding in the French cohort was the presence of dysregulated T Lymphocytic function seen in 23% of the cases (10/43).
Histopathological findings are overall quite limited.4, 29 The China Cohort included four patients who underwent brain biopsy and the pathological examinations revealed substantial inflammation and prominent perivascularly located B and T cells. 4 Immunohistochemical analysis revealed CD20+ B cells, CD3+ T cells and CD138+ antibody-producing cells, the latter of which were most abundant in the Virchow robin spaces. Additionally, there was a total loss of AQP4 and GFAP in one patient and focal reduction of GFAP and AQP4 was found in three other patients. 4 Unlike Aquaporin 4-associated disease, GFAP astrocytopathy is associated with neuronal loss in the affected areas. The mechanism by which antibodies to an intracellular antigen caused such profound inflammation remained unclear. The accepted hypothesis for this phenomenon is that these antibodies, rather than being directly pathogenic, serve as a surrogate marker for cytotoxic T-cell-mediated inflammatory response. 4
During the evaluation of cases presenting with subacute neurological syndrome, the question of Infection versus inflammation is often a difficult one to answer. Infections like tuberculosis mimic all commonly encountered symptoms in underlying neuroinflammation and misdiagnosis may occur.5, 30 In such cases, isolation of the GFAP antibody can help make therapeutic decisions emergently when time is scarce as presence of anti-GFAP antibodies in the CSF is highly specific.2, 3, 16 Other clinical mimics where isolation of GFAP antibody is of diagnostic relevance include anti-aquaporin positive NMOSD, 14 myelin oligodendrocyte glycoprotein associated disease (MOGAD), 31 chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids (CLIPPERS)14, 32, 33 and CNS lymphoma. 34 In addition, isolation of this antibody will also prompt screening for any underlying malignancy which in turn can make an early diagnosis.
Additional avenues that have to be explored include identifying a biomarker to predict relapses thus facilitating early initiation of immunomodulators. When repeated after an interval, 11 among 22 cases of the French Cohort tested negative. 3 Whether serially testing antibodies to monitor the disease activity will reduce the risk of a relapse is yet unknown. Furthermore, while a diagnostic flow chart has been formulated by Huang et al., 35 uniform diagnostic criteria are yet to be established. There is an ongoing ambispective study that will likely provide further clarity on the disease. 36
Conclusion
Autoimmune GFAP astrocytopathy encompasses an expanding clinical spectrum and should be considered in the context of myelitis, optic neuritis, ataxia, papillitis, seizures autonomic dysfunction, peripheral neuropathy and movement disorders occurring in isolation or more commonly in varying combinations. The detection rate of this entity is increasing and GFAP astrocytopathy was noted to be present in 5% of patients with suspected autoimmune disease. 5 Awareness of this disease is of paramount importance in a resource-limited setting where entire panels of antibodies cannot be run and clinical pattern recognition is of utmost relevance. The first patient remained undiagnosed despite undergoing extensive evaluation and having all the key features of the disease. The second patient was thought to have a purely nonorganic aetiology for her symptom complex. The third and fourth patients remained in remission while on rituximab, highlighting the efficacy of B cell-depleting agents in relapsing-remitting neurological diseases. Exquisite steroid responsiveness was noted in all of our four cases.
Our case series shows a favourable clinical profile and the primary hurdle encountered in all four cases was to establish a diagnosis, further stressing the need for a predictive diagnostic algorithm.
Defining and validating uniform diagnostic criteria, identifying prognostic factors and determining optimal therapeutic strategies in GFAP astrocytopathy are the major issues that have to be addressed in the coming years.
Footnotes
Acknowledgements
No person who had contributed substantially to the production of this article had been excluded from authorship. Persons who have contributed partially have been acknowledged in the article.
Authors’ Contribution
Contributor 1: Conception and design of the work, data acquisition, statistical analysis, and interpretation of data, literature review manuscript preparation and editing.
Contributor 2: Conception and design of the work, data acquisition, statistical analysis, and interpretation of data, literature review manuscript preparation and editing.
Contributor 3: Conception and design of the work, analysis, interpretation of data, manuscript editing and review.
Contributor 4: Data analysis, interpretation of data, manuscript preparation and editing.
Contributor 5: Data acquisition, data analysis, and interpretation of data and manuscript preparation.
Contributor 6: Data acquisition.
Contributor 7: Data acquisition, manuscript preparation.
Data Availability
All data will be made available by the corresponding author on request.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
ICMJE Statement
This article has been edited in accordance to the guidelines of the International Committee of the Medical Journal Editors.
Statement of Ethics
We the authors state that subjects have given their written informed consent and that the study protocol was approved by the institute’s ethical committee (Father Muller institutional ethics committee FMIEC Ref ID489/2023).
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.