
Abstract
Background
Epilepsy is a chronic neurological disorder that affects approximately 50–70 million people worldwide. Epilepsy has a significant economic and social burden on patients as well as on the country. The recurrent, spontaneous seizure activity caused by abnormal neuronal firing in the brain is a hallmark of epilepsy. The current antiepileptic drugs provide symptomatic relief by restoring the balance of excitatory and inhibitory neurotransmitters. Besides, about 30% of epileptic patients do not achieve seizure control. The prevalence of adverse drug reactions, including aggression, agitation, irritability, and associated comorbidities, is also prevalent. Therefore, researchers should focus on developing more effective, safe, and disease-modifying agents based on new molecular targets and signaling cascades.
Summary
This review overviews several clinical trials that help identify promising new targets like lactate dehydrogenase inhibitors, c-jun n-terminal kinases, high mobility group box-1 antibodies, astrocyte reactivity inhibitors, cholesterol 24-hydroxylase inhibitors, glycogen synthase kinase-3 beta inhibitors, and glycolytic inhibitors to develop a new antiepileptic drug.
Key messages
Approximately 30% of epileptic patients do not achieve seizure control. The current anti-seizure drugs are not disease modifying, cure or prevent epilepsy. Lactate dehydrogenase inhibitor, cholesterol 24-hydroxylase inhibitor, glycogen synthase kinase-3 beta inhibitors, and mTOR inhibitors have a promising antiepileptogenic effect.
Introduction
Epilepsy is a chronic, usually life-long neurological disorder characterized by the periodic and unpredictable occurrence of unprovoked seizures. Seizures occur when a neuron (mostly in the cortex) induces synchronous, abnormal, paroxysmal, and recurrent discharges. Epilepsy is the most prevalent central nervous system (CNS) disorder that affects 0.5%–1% of people in the world. 1 The pathophysiology of epilepsy involves distortion of the normal balance between glutamate-stimulating neurotransmitter and γ-aminobutyric acid (GABA)-inhibitor neurotransmitter, deposition of epileptic insults, and changes in neural circuits that over-synchronize neurons. Decreased GABA and increased glutamatergic signaling pathways cause several types of epilepsy. 2 Several pathophysiologic insults cause epilepsy at different levels of CNS activity, such as neuronal structure change following prolonged febrile seizures, neurotransmitter synthesis, synaptic development, and abnormal ionic channel and receptor activity. Several neuronal activities, like neuronal circuit changes, abnormal dendrite structure, minimized GABA production, abnormal GABA receptors, and increased glycine synthesis, cause overstimulation of glutamate receptors.2, 3
Antiepileptic drugs (AEDs) suppress the abnormal activity of neurons called epileptic discharges. AED may also have neuroprotective potential. The proposed mechanisms of action of AEDs are modulation of calcium, sodium, or potassium voltage-dependent channels, increased synaptic pathways by GABA, and decreased glutamate-induced stimulation. Most AEDs are metabolized in the liver, and some drugs are metabolized into active metabolites. 4
Patients with seizures face physical, psychological, and social morbidities. These difficulties can be prevented by controlling seizures. Eliminating seizures is the most important objective of epilepsy management. So, most AEDs help to control the occurrence of seizures. The efficacy of AED monotherapy is well established, and about 60%–70% of patients effectively control their seizures. 5 Switching to other AED monotherapy controls half of the remaining 30% of drug-resistant patients. The remaining patients effectively controlled the seizures by using a combination of AEDs. 6 Polytherapy with AEDs causes drug interactions and exacerbates the side effects. Regardless of the progress attained in management, the mortality of patients due to epilepsy has not decreased. Thus, it is clear to search for new and novel AEDs for the treatment of epilepsy. 7 Most AEDs need serum concentration measurements for proper management of epilepsy due to their narrow therapeutic range property. Secondly, the narrow therapeutic index of AEDs causes serious idiosyncratic side effects after weeks of starting the AED treatment. Besides, cumulative toxicity is another side effect that occurs after a long period of epilepsy treatment.8, 9 Therapeutic use of AEDs during pregnancy increases the risk of congenital abnormalities.
Administration of valproic acid in the first trimester increases the rate of major anomalies, such as spina bifida, cardiac, craniofacial, skeletal, and limb defects, and decreases intrauterine growth. Carbamazepine causes an increased risk of fetal abnormalities such as spina bifida, and phenytoin causes major malformations. 10 Even if the adverse drug reaction and pharmacokinetic character of the recent AED are improved, the treatment of epilepsy still depends on drugs that control the symptoms but do not hinder the disease’s mechanism. To minimize these clinical challenges, understanding the molecular mechanisms of epileptogenesis will pave the way to develop new drugs with broad therapeutic use that can potentially control the disease’s development or improve its clinical outcome.10, 11 After several decades of research, many drugs were approved with better absorption, distribution, metabolism, excretion, and side effects relative to the first generation of AEDs. However, not enough progress has been made in terms of efficacy in drug-resistant epilepsy. This fact demands the conscientious strategies used to develop anticonvulsant agents. 12
Mechanisms of Action of Existing AEDs
The current therapies decrease neuron firing by acting on ion channels that produce action potentials or on receptors that regulate synaptic transmission. 13 The newer drugs have novel mechanisms and better safety profiles. This choice of AEDs has made it possible to tailor treatment plans based on patient profiles. Despite an increase in the choice of drugs, we still treat only the symptoms of seizures without arresting the underlying causes of epileptogenesis or providing neuroprotection from seizures. 14 The mechanisms of action of AEDs include voltage-dependent ion channel blockade, increasing inhibitory neurotransmission, and suppressing excitatory neurotransmission, as shown in Figure 1.
The Mechanisms of Action of the Antiepileptic Drugs.
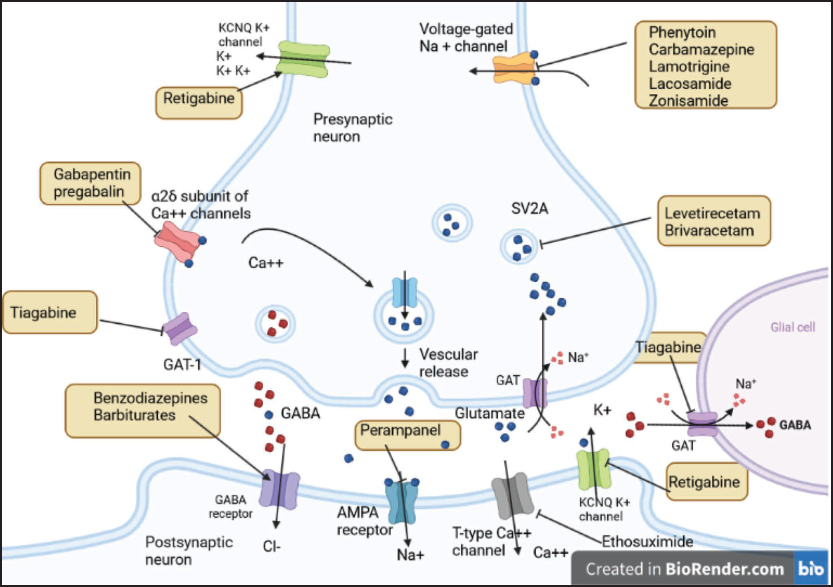
Voltage-dependent Sodium Channel Blockers
Voltage-dependent sodium channel (VGSC) blockers have been the major AED for the management of focal and generalized tonic-clonic seizures for more than 70 years. VGSCs are one of the vital targets for the management of several CNS diseases, such as epilepsy, psychiatric disorders, chronic pain, and spasticity. VGSC blockers play a key role in the induction and propagation of action potentials in neurons. This reduction in cellular excitability helps to improve the symptoms of epileptic conditions. Most AEDs act by inhibiting sodium channels. 15 AEDs such as phenytoin, carbamazepine, and valproate bind to the active state of the channel and reduce high-frequency firing during a seizure; some of the newer AEDs, including lamotrigine, oxcarbazepine, and zonisamide, act by facilitating the fast inactivation of sodium channels. Besides, lacosamide and eslicarbazepine increase the slow inactivation of VGSCs, which may be associated with better tolerability at higher dosages. 16 VGSC exists in three states: resting, open, and inactivated. Most AEDs with VGSC blockers have a high affinity for the inactivated state. As a result, these drugs produce a voltage- and frequency-dependent reduction in channel conductance, resulting in a limitation of repetitive neuronal firing with little effect on the generation of single action potentials. 17
Voltage-Gated Calcium Channel Inhibitor
Calcium ions enter via voltage-activated calcium channels (VGCC) and depolarize the cell membrane to control intracellular signaling. VGCCs are grouped into two main classes based on the threshold of action potential: low VGCC (T-type) and high voltage-activated (HVA) calcium channels (L-, N-, P-, Q-, and R-type). Each VGCC is composed of α1, α2, δ, β, and γ subunits. While L-type VGCC plays a key role in the excitation of postsynaptic neurons, N-, P/Q-, and R-type VGCC are involved in rapid synaptic neurotransmitter release. 18 VGCC is a key component in controlling neuronal excitability and is thus of importance in the pathogenesis of different types of epilepsies, including absence epilepsies, which account for 10% of epileptic seizures.18, 19
T-type calcium channels initially respond to small depolarization, and their effect further depolarizes the cell, where HVA and sodium channels activate, allowing excess calcium entry and action potential generation. 20 VGCC is an important target for most of the AEDs. Ethosuximide is the first-line drug for the management of absence epilepsy and acts by inhibiting the T-type calcium channel. Sodium valproate, lamotrigine, zonisamide, phenobarbital, felbamate, gabapentin, pregabalin, and topiramate also inhibit calcium channel conductance. Several AEDs cause adverse events, but ethosuximide has fewer side effects. Ethosuximide’s most prevalent side effects are gastrointestinal and neurological. Drowsiness may occur at the beginning of treatment but is resolved with dose reduction. Ethosuximide is more toxic in pregnant women with epilepsy than in controls.21, 22 Currently, ACT-709478 (NBI-827104) is a T-type VGCC blocker in phase II studies to treat a rare form of pediatric epilepsy and essential tremor.23, 24
GABAA Receptor Modulator
GABAA receptor is a ubiquitous ligand-gated ion channel that possesses five components (α, β, γ, δ, ε, or ρ) that can be activated by GABA binding. This arrangement causes a different assortment of receptor subtypes in most parts of the CNS. There are several allosteric binding sites at the GABA A receptor. The GABAA α1 mediates sedating and amnesic activity; α2 receptors mediate anxiolytic activity; α subunits are involved in the anticonvulsant effect, α5 mediates learning and memory; and β3 subunit deficiency decreases GABA inhibition.25, 26
Several studies reported that the full GABAA receptor agonist has great potential to develop physical dependence and tolerance. Besides, partial positive allosteric modulators possess a significant effect on drug-resistant epilepsy. Therefore, partial positive allosteric modulators of GABAA receptors are crucial for the management of drug-resistant epilepsy. Most AEDs approved for the management of epilepsy act by activating GABAA receptors, enhancing the inhibitory effect of GABA.26, 27
Barbiturates and benzodiazepines activate the GABAA receptor to open the chloride ion channel. Barbiturates prolong the chloride channel opening duration, but benzodiazepines increase the chloride channel opening frequency. Phenobarbital activates the GABAA receptor in the absence of GABA but not benzodiazepines.27, 28 Felbamate and topiramate also modulate GABA responses at the GABAA receptor. Levetiracetam indirectly influences GABAA receptor function by reducing the negative allosteric modulation of the receptor. Vigabatrin is an irreversibly inhibited GABA-transaminase that metabolizes GABA, whereas tiagabine inhibits the reuptake of GABA from the synaptic cleft by inhibiting GABA transport. 29 Stiripentol and clobazam are selective positive allosteric modulators. GABAA enhancers selectively bind to α3-B3-γ2 and alpha2-subtype GABAA receptors, respectively. Several selective GABA A receptor agonists have the chance to be effective against specific types of epilepsy.30, 31 Several compounds are in clinical trials, including ganaxolone (phase III), 31 CVL-865 (phase II), and padsevonil (phase III). 32
Glutamate Receptor Blocker
Glutamate is the principal fast excitatory neurotransmitter in the CNS. Glutamate receptors respond to glutamate binding by increasing cation conductance, resulting in neuronal excitation. Glutamate receptors are grouped as ligand-gated ion channels (“ionotropic”) and “metabotropic” GPCRs. The ionotropic receptor has three subfamilies designated as α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA), kainate, and N-methyl-
Felbamate blocks the NMDA subtype of glutamate receptors. Topiramate, lacosamide, and zonisamide exert their effects by inhibiting AMPA and kainate receptors. Phenobarbital blocks AMPA receptors. Lamotrigine selectively reduces glutamate release by inhibiting presynaptic sodium and calcium channels rather than having a direct effect on the glutamate system.38, 21 Several AMPA receptor antagonist agents are in clinical development for refractory status epilepticus, including NS-1209 and talampanel (GYKI-53773). 39 In January 2021, the FDA approved a phase IIa study of the selective metabotropic glutamate type 2 (mGlu2) receptor as a positive allosteric modulator JNJ-40411813 (ADX71149) in epilepsy patients.39, 40
Synaptic Vesicle Protein 2A
Synaptic vesicles exist in the presynaptic nerve and are used to store and regulate exocytosis. Synaptic vesicle protein 2A (SV2A) is a synaptic vesicle that is found in all kinds of nerve terminals. 41 SV2A protein expression decreased by 40% in brain tissues resected from patients with temporal lobe epilepsy-resistant drugs. 42 Levetiracetam, brivaracetam, seletracetam, and padsevonil are new groups of AED that bind to a presynaptic vesicle protein called SV2A, causing synaptic vesicles to fuse with the presynaptic membrane and release neurotransmitters. 43 These drugs also inhibit presynaptic voltage-gated Ca2+ channels and inhibit the release of Ca2+ from intracellular stores, both of which suppress excitability and reduce transmitter release. SV2A inhibitors are used as adjunctive management for drug-refractory partial-onset seizures in patients with epilepsy aged 16 years and above. Brivaracetam is more selective and has a higher affinity for SV2A than levetiracetam.43, 44
KCNQ/Kv7 Potassium Channel Opener
Voltage-gated K+ channels (Kv) play a crucial role in controlling intrinsic electrical properties in excitable cells. Kv channels have a KV7 family that includes five members (Kv7.1-5) who have gained great interest in developing a novel drug due to its link to different diseases. KCNQ1 is located in cardiac tissues, whereas KCNQ2–5 is mainly found in the CNS and is therefore referred to as a neuronal KCNQs. 45 Kv7.2 and Kv7.3 subunits assemble into KCNQ2/3 heterotetramers, which are identified as a major part of the low voltage-dependent M-channels that control neuronal activation. Facilitating activation or delaying inactivation of the Kv7 channel suppresses neuronal excitability, which helps to control electrical activity during seizures. 46 The opening of potassium channels causes hyperpolarization, which causes a reduction in excitability. Retigabine is the first AED approved for the add-on treatment of refractory focal epilepsy. 47 It acts as an opener of KCNQ channels that selectively acts on neuronal Kv7 potassium channels, but now it is not commercially available due to its side effects. Retigabine may cause urinary retention due to potentiating KCNQ channels expressed in bladder tissues. However, reports of skin discoloration on peripheral tissue and loss of vision due to retinal discoloration eventually led to a black box warning on retigabine medication.47, 48 XEN1101 is another novel positive allosteric modulator of KCNQ2/3 currently being developed by Xenon in phase II clinical trials for the treatment of focal epilepsy. 49
Carbonic Anhydrase
The acid-base balance and maintaining local pH are essential for the normal activity of the CNS. A seizure can be caused by an increase in the extracellular potassium level or by neuronal excitability affected by pH changes. Generally, the excitability of neurons is enhanced by alkalosis and suppressed by acidosis. 50 Carbonic anhydrases are ubiquitous enzymes that have a key role in catalyzing the bidirectional conversion of carbon dioxide and water to bicarbonate and hydrogen ions. (CO2 + H2O ↔ HCO3- + H+)
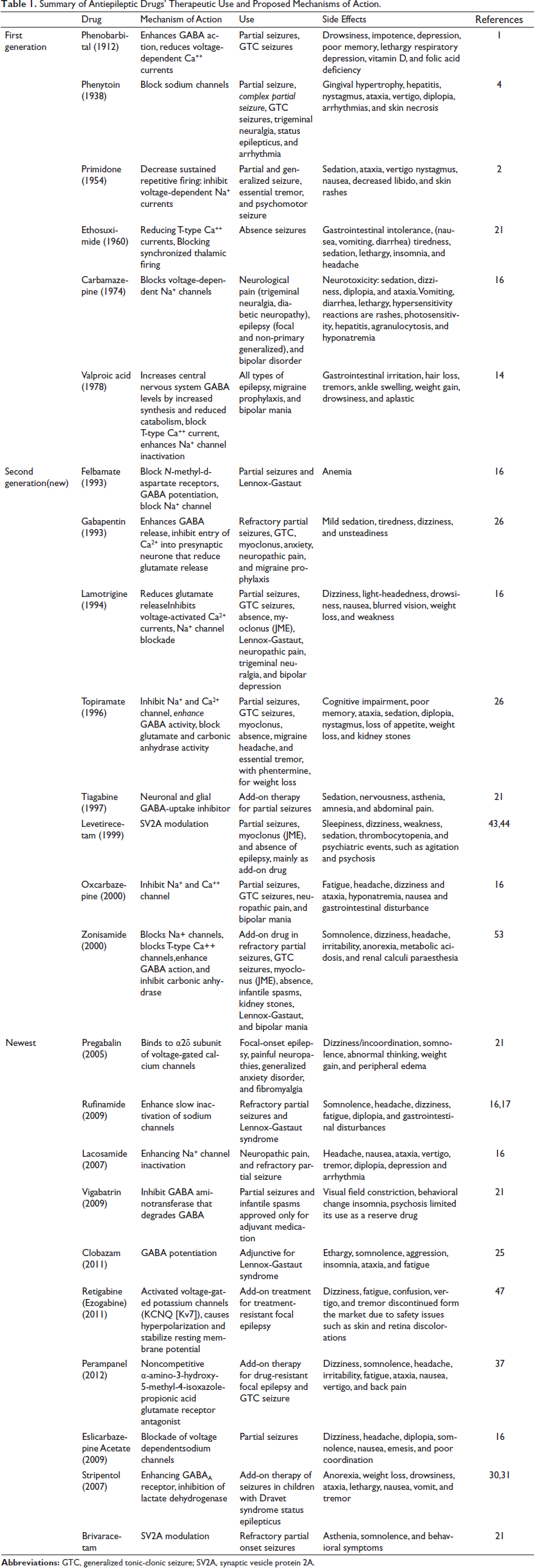
Carbonic anhydrase inhibitors such as acetazolamide, methazolamide, topiramate, zonisamide, and sulthiame can reduce seizures. These drugs act by producing localized acidosis and increasing intercellular hydrogen ion concentrations.50, 51 This causes the shift of potassium ions to the extracellular space and causes hyperpolarization. This suppresses excitatory neurotransmission by reducing NMDA receptor activity and enhances inhibitory neurotransmission by facilitating the responsiveness of GABAA receptors. Acetazolamide is a carbonic anhydrase inhibitor used as an AED with some success. 52 Topiramate and zonisamide inhibit carbonic anhydrase but are less potent. Thus, carbonic anhydrase inhibition can be proposed as an AED mechanism, but the extent to which it contributes to the clinical effect remains to be determined. The summary of AEDs is presented in Table 1.52, 53
Summary of Antiepileptic Drugs’ Therapeutic Use and Proposed Mechanisms of Action.
Novel Targets for Antiseizure Therapy
Currently, AED development has a better chance due to a better understanding of the mechanisms of epileptogenesis, seizure generation, pharmacoresistance, and the presence of disease-specific models. Several studies on the mechanisms of seizure initiation and propagation have identified several promising novel targets for developing drugs for the treatment of epilepsy. AEDs that inhibit epileptogenesis are urgently needed. The advancement in preclinical and clinical trials brings hope for developing a new agent with a novel target that has better efficacy and safety. 54 In recent years, there have been numerous successes in AED discovery, but still, treatment of epilepsy is challenging, and safe and effective AEDs are far away. 55 Targeting the underlying mechanism of epileptogenesis helps to develop AED with few side effects. For the past 20 years, a new generation of AEDs has been approved for the treatment of different types of epilepsy. 56 Most of the recently approved AEDs were modifications of the available AEDs with better adverse effects and stability. These new AEDs are used either alone or added to other drugs to treat refractory epilepsy. Nowadays, several novel antiepileptic agents are in different drug development stages.56, 57 These drugs may have better safety, efficacy, and compatible pharmacokinetic properties. It is urgent to improve animal models and search for new drug targets to treat patients with refractory epilepsy.57, 58
Lactate Dehydrogenase Inhibitor
Several studies have reported that glycolysis is vital for maintaining sustainable synaptic activity. To support the enhanced synaptic vesicle effect, significant glycolysis is undertaken in activated neurons. 59 Lactate dehydrogenase (LDH) catalyzes the interconversion of lactate and pyruvate; the latter is the final metabolic product of glycolysis. Lactate is used as fuel by several tissues during the aerobic state. In the CNS, lactate is the main source of energy. 60 The concept of the astrocyte-to-neuron lactate shuttle hypothesis is that astrocytes produce lactate, which is then transported into neurons. Then, the transported lactate is metabolized to pyruvate by LDH, which helps to produce energy in the Krebs cycle. Based on this hypothesis, LDH inhibitors suppress neuronal discharges and play a key role in inhibiting epilepsy. 61 Oxamate is a pyruvate analog that inhibits the LDH enzyme. A study done on the administration of Oxamate into the hippocampus inhibits seizures in animal models of temporal lobe epilepsy. Finally, the authors propose that some currently used AEDs may inhibit LDH.59, 62
Among 20 AEDs screened on purified LDH enzyme, only stiripentol showed a partial LDH inhibitory effect at the level of 500 µM. 63 Stiripentol has antiepileptic activity through two mechanisms. The first mechanism inhibits coadministered AED metabolism, and the second mechanism is GABA receptor modulation. These mechanisms may contribute to the treatment of Dravet syndrome. 64 The mammalian target of rapamycin (mTOR) signaling pathway is also involved in epilepsy and is inhibited by the LDH inhibitor oxamate. Activation of the mTOR pathway enhances LDH expression. Furthermore, rapamycin, an mTOR inhibitor, decreases LDH expression, reduces lactate concentration, and inhibits epileptic seizures. Therefore, compounds with LDH inhibitory activity may contribute to the development of new AEDs for the management of drug-resistant epilepsies. 65
c-Jun N-Terminal Kinases
Mitogen-activated protein kinases (MAPKs) are highly conserved serine (Ser)/threonine (Thr) kinases that control the proliferation, differentiation, senescence, and apoptosis of cells. These signals are transmitted by four signal cascades, including kinesis (ERK) 1/2, ERK5, p38, and the c-Jun N-terminal kinases (JNKs), to modulate the activity of transcription factors, transcription suppressors, and chromatin remodeling proteins. 66 JNKs are also called stress-activated protein kinases, which are stimulated by cytokines, oxidative stress, DNA-damaging agents, UV radiation, protein inhibitors, and growth factors. JNK has an essential role in cellular proliferation, differentiation, morphogenesis, and the maintenance of synapses. 67 Besides, it regulates neuronal migration and death via apoptosis after cellular insults. These activities suggest JNK has a key role in psychiatric and neurodegenerative disorders. 67
Tai and colleagues reported significant JNK hyperactivation in rats treated with pilocarpine hydrochloride. 67 In another study, rats treated with anisomycin, a nonspecific MAPK activator, increased seizure frequency. 68 To confirm the hypothesis that JNK overactivation could be relevant in chronic epilepsy. JNK inhibitor (SP600125) showed a significant reduction in convulsive seizure frequency. 69 In the pilocarpine-induced rat model of epilepsy, elevated phosphorylated JNK concentrations indicated JNK hyperactivation in the CA1 region of the hippocampus, and SP600125, a JNK inhibitor, displayed a reduction in seizure occurrence.67, 70
High Mobility Group Box-1 Antibody
Our body has mechanisms to identify pathogenic agents through the detection of substances such as damage-associated molecular patterns (DAMPs). DAMPs such as S100 proteins, high mobility group box-1 (HMGB1), and heat shock proteins are endogenous danger molecules released by damaged tissue. DAMPs are vital during tissue repair and regeneration and in autoimmune disorders. The HMGB1 nuclear protein is the only DAMP that induces innate immunity either by itself or via cytokines. 71 HMGB1 triggers autophagy when transported into the cytoplasm. HMGB1 is a proinflammatory cytokine that triggers status epilepticus and febrile seizures. 72
In epileptic patients’ brains, high amounts of HMGB1, RAGE, TLR4, and nuclear factor-kappa B (NF-κB) were reported. The increased amount of HMGB1 is related to the onset of seizures, confirming HMGB1 as a contributor to neuron hyperexcitability, which is critical in seizure onset. Hence, the HMGB1/RAGE/TLR4 pathway may initiate the development and persistence of seizures. 73 The translocation of HMGB1 was reported in different animal models such as pentylenetetrazole (PTZ)-induced seizures, pilocarpine-induced status epilepticus, and KA-induced epilepsy. 74 A monoclonal antibody that targets HMGB1, also called “anti-HMGB1 mAb,” decreases inflammation in the CNS, which may inhibit epileptogenesis and seizures in animal models. Anti-HMGB-1 mAb showed rapid anti-seizure activity in maximal electroshock and PTZ-induced seizures in mice models. 75 Mice treated with anti-HMGB-1 mAb for 2 weeks did not show drug resistance and had a wide therapeutic index that provided a chance of using large doses without side effects.75, 76
Astrocyte Reactivity
Several studies showed that the leakage of serum albumin via a disrupted blood–brain barrier (BBB) may initiate specific signaling cascades within neurovascular cells that specifically activate astrocytes. These reactive astrocytes release glutamate, resulting in neuron excitability and epileptogenesis. 77 Besides, reactive astrocytes release proinflammatory cytokines (e.g., interleukin [IL]-1β, IL-6, and tumor necrosis factor-α) and recruit more inflammatory cells by secreting C-C motif chemokine ligands 2, 3, and 5. As a result, there is increased neuronal excitability, seizure development, neuronal inflammation, and death. 78
Albumin binds to astrocytic transforming growth factor-beta (TGF-β)R2 resulting in the activation of the TGF-β signaling pathway, synthesis of TGF-β, and astrocyte activation that inhibits buffering of potassium and glutamate at the cellular level. ALK5/TGF-β-pathway induces excitatory synaptogenesis, whereas SJN2511, specific ALK5/TGF-β inhibitors, prevents synaptogenesis and epilepsy. 79 Hence, TGF-β pathway inhibition arrests astrocyte activation during epileptogenesis, leading to a reduction in seizures and brain inflammation.78, 79 So, the TGF-β pathway inhibition may serve as a therapeutic target to prevent seizure development in individuals with brain injury. The excitatory/inhibitory balance is controlled by astrocytes, and its disruption may cause neural cells to become epileptic. Thus, activation of adenosine type-1 receptors interferes with glutamate release in astrocytes, which has an anti-seizure effect. 80
Cholesterol 24-Hydroxylase Inhibitor
Cholesterol plays an essential role in maintaining different physiological activities in the CNS, such as controlling membrane potential and the release of synaptic vesicles. The regulation of cholesterol metabolism in the CNS differs from that in the periphery. One such example is the cholesterol 24-hydroxylase (CH24H) enzyme, which is a CNS-specific enzyme that metabolizes cholesterol into 24S- hydroxycholesterol, the main mechanism of cholesterol clearance from the CNS. 81
Modulation of CH24H affects amounts of 24- hydroxycholesterol, which is a positive allosteric modulator of NMDA receptors in the CNS. Overactivation of NMDA receptors causes neuronal cell death and epilepsy. 82 Besides, neuronal 24-hydroxycholesterol may have a crucial role in controlling glial cell activity, including inflammation, oxidative stress, and potassium homeostasis. So, CH24H inhibition may be important in epilepsy. 83 Soticlestat (TAK-935) is a promising therapeutic agent for the treatment of epilepsy. TAK-935 decreased the concentration of 24-hydroxycholesterol in the CNS as reported in an animal model. Currently, soticlestat (TAK-935) is in a phase Ib/IIa clinical trial as adjunctive therapy in pediatrics with Dravet syndrome and Lennox-Gastaut syndrome. Soticlestat was well tolerated and reduced seizure frequency during the study period.83, 84
Glycogen Synthase Kinase-3 Beta and Tau Protein
Glycogen synthase kinase-3 (GSK3) is a protein-directed kinase mostly found in axons, a highly conserved Ser/Thr kinase that controls several biological processes such as immunity, metabolism, cell differentiation, and survival. GSK3 is involved in the pathogenesis of several neurological disorders such as schizophrenia, bipolar disorder, Parkinson’s disease, brain tumors, stroke, and Alzheimer’s disease. 85 Overexpression of GSK3β facilitates tau protein phosphorylation, microtubule dissociation, and aggregation that causes axonal transport changes, neurodegeneration, and impairment of learning and memory. Tau is a microtubule-associated protein that stabilizes microtubules in the axon by interacting with kinesin and dynein proteins. 86 The storage of hyperphosphorylated tau protein causes excitatory and inhibitory neurotransmitter imbalances that disrupt neuronal synchronization and epilepsy development. Patients with cognitive impairment and epilepsy showed aggregation of hyperphosphorylated tau protein and enhanced GSK3β enzyme activity.85, 87
Several studies showed that decreased tau concentration has a preventive effect against seizures and halts neuronal and synaptic loss. In epilepsy, inhibition of GSK3β reduces tau hyperphosphorylation and neurofibrillary tangle development. TDZD-8 is the first non-ATP-competitive inhibitor of GSK3β. TDZD-8 inhibits tau hyperphosphorylation, neuronal death, and the modification of neurogenesis in epileptic patients. 88 Ifendropil and memantine are NMDAR blockers that have GSK3β inactivation and reduce tau phosphorylation. 89 Recent studies showed that GSK3β inhibitors such as indirubin and BIO-acetoxime have anticonvulsant effects in PTZ-induced seizures in zebrafish and mouse 6 Hz model.88, 89
Glycolytic Inhibition
A ketogenic diet is rich in fat (90%), with a low amount of carbohydrates and protein. So our body uses fat as a source of fuel, producing ketone bodies and resulting in glycolysis reduction. Glycolysis inhibitor mimics a ketogenic diet that helps to control the seizure. The glycolytic inhibitor 2-deoxy-
2-DG is a glycolysis inhibitor that showed promising seizure protection. It inhibits glycolysis by phosphorylation of 2-DG at the sixth position, which interferes with the isomerization of glucose-6-phosphate to fructose-6-phosphate. This decreases glycolytic flux, reduces energy production, and produces antiseizure effects. 2-DG showed a neuroprotective effect by decreasing the deleterious oxidative and metabolic effects of kainic acid in hippocampal cell cultures. 2-DG also decreased the seizure-induced hippocampal loss in kainic acid-induced seizures in animal models. 92 The neuroprotection effect of 2DG may be associated with the inhibition of phosphoglucose isomerase, resulting in disruption of glycosylation, increased tonic GABA inhibition by increasing neurosteroidogenesis, activation of adenosine monophosphate kinase, and reduced oxidative stress. During seizures, a high amount of energy is needed for the neurons, and this energy is gained by increasing glycolysis. But the presence of 2-DG decreases NADH amounts, and the kindling process is decreased.92, 93
Inhibition of mTOR Pathway
The mTOR is a serine/threonine protein kinase that regulates cell division, differentiation, transcription, translation, proliferation, and survival. Abnormal mTOR signaling contributes to different disorders, including cancer, diabetes, and epilepsy. mTOR signaling cascade induction is associated with epileptogenic diseases called “mTORpathies,” which are characterized by pharmacoresistant epilepsy and altered neuronal morphology because of mutations resulting in mTOR pathway activation. mTORC1 activation upregulates AMPA receptor activity in vitro. 94 Hyperactivation of the mTOR cascades contributes to epileptogenesis in an animal model. The mTOR inhibitors have displayed antiepileptogenic activity in animal models of epilepsy. Rapamycin is an inhibitor of mTOR that prevents epilepsy via suppression of astrocyte and microglial cell activation. 95 Rapamycin controls seizures and improves behavioral impairments by inhibiting IL-2 and IL-1β linked activation of PI3K/AKT/mTOR signaling in animal models of temporal lobe epilepsy. Management of epilepsy with everolimus decreased seizure frequency and severity. 89 Everolimus is a rapamycin analog approved for the treatment of refractory seizures in 2017. But rapamycin and its derivatives have poor oral bioavailability, less BBB entrance, and slow accumulation in the CNS.
Several studies showed that mTOR inhibitors are better for early preventative therapy of epilepsy than the standard drug-resistant epilepsy treatment.96, 97 Recently, two selective mTORC1/C2 inhibitors named PQR620 and PQR530 were developed. PQR620 showed better dose-dependent antiseizure efficacy. Resveratrol, a natural compound, has antiepileptogenic activity by suppression of NF-κB and proinflammatory cytokine synthesis that is affected by mTOR signaling. The ketogenic diet has an anti-inflammatory effect and also blocks mTOR activity.90, 98
G Protein-Coupled Estrogen Receptor 1
The genomic modulation of neuronal activity acts via its ERα and ERβ receptors. G protein-coupled estrogen receptor 1 (GPER1) is a novel estrogen receptor that mediates the non-genomic neuroprotective effects of estrogen in brain injury, cerebral ischemia, Alzheimer’s disease, and Parkinson’s disease. Besides, GPER1 involve in cognition, depression, homeostasis, pain, and other neuronal activities. 99 GPER1 is expressed in synaptic and extrasynaptic regions within dendritic spines. In the dorsal hippocampus, GPER1 facilitates social recognition and object placement performance. The neuroprotective effects of 17β-estradiol in epilepsy, Alzheimer’s, and Parkinson’s disease are mediated through GPER1 and ion channels. 99 Estrogen can protect the hippocampus neurons against seizure-induced damage and prolong seizure latency. Besides, 17β-estradiol has a neuroprotective effect against the excitotoxicity damage associated with seizures and inflammation. The knockdown of the estrogen receptor showed overexpression of proinflammatory cytokines and reactive microglia that indicated enhanced inflammation post-SE.100, 101
The Endocannabinoid System
The endocannabinoid system (ECS) is involved in the control of excitatory and inhibitory synaptic transmission in the CNS. ECS has two G protein-coupled receptors, the cannabinoid receptor (CB1R) and the CB2R, with two endogenous cannabinoid ligands, namely 2-arachidonoylglycerol (2-AG) and N-arachidonoylethanolamide (NAN), respectively.
The presence of these compounds at the synaptic site modulates these cells’ activity. 102 The major CB1R in the CNS is CB1R, a presynaptic G-protein-coupled receptor that activates VGCCs and enhances potassium-channel conduction in the presynaptic neurons. CB1R is activated by the activity-dependent synthesis of 2-AG and is involved in the retrograde control of synaptic transmission. 103
Several studies have shown that ECS insults contribute to epilepsy development. Patients with temporal lobe epilepsy have lower anandamide levels in the CNS, lower levels of CB1R messenger RNA, and reduced expression of diacylglycerol lipase α (DAGL-α), the enzyme that helps for the synthesis of 2-AG in postsynaptic neurons.102, 103 Another study showed that epileptic patients show increased expression of CB1 receptors on astrocytes. In mice, activation of astrocytic CB1 receptors caused glutamate release from astrocytes, suggesting that the elevated CB1 receptor expression in astrocytes observed in epileptic patients may increase the excitability of the neuron. These studies support the suggestion that the ECS has a key role in the inhibition of seizures in patients with epilepsy. Upregulation of CB1R activity has an antiseizure effect; reducing the degradation of endocannabinoids ameliorates experimentally induced seizures. 104 CB2Rs have low expression in the CNS in a normal state but are inducible during CNS disorders (epilepsy) and appear to be crucial for neuroprotection. Targeting CB2Rs may also provide a novel treatment approach for managing epilepsy without CB1R-mediated side effects. Several studies showed that CB2Rs were involved in epileptic activity in animal models. 105
In the PTZ animal model, pretreatment with palmitoylethanolamide (PEA) increased the latency of seizures and decreased the duration of seizures, and this antiseizure activity was decreased by the CB2R antagonist (AM630), suggesting that CB2Rs mediate PEA’s effect. Huizenga et al. studied the antiseizure activity of different cannabinoid compounds and found that either combined CB1R/CB2R or selective CB1R agonists have antiseizure activity. 106
Even though the selective CB2R agonist HU-308 did not display antiseizure activity, the selective CB2R antagonist AM630 increased seizure severity. Besides, CB1R knockout mice did not show an epilepsy phenotype, but both CB1R and CB2R knockout mice showed epilepsy, suggesting that CB2Rs activation suppresses activated neurons and inhibits epileptic seizures. 102 WIN 55212-2, a non-selective CB receptor agonist, has antiseizure activity in epileptic models. 107 CB2R knockout mice showed increased seizure occurrence, suggesting that CB2Rs increase seizure susceptibility. Caryophyllene, a CB2R agonist, improved seizure effects in mice models. Generally, the activation of CB2Rs has an antiseizure effect. Although several studies showed that CB2R agonists suppress different epileptic seizures, they suggest that CB2R is a promising therapeutic target for treating epilepsy. 102 Phytocannabinoid agents significantly decreased the severity of seizures and post-seizure mortality in animal models. Epidiolex is a plant-derived cannabidiol oral solution approved by the FDA to treat seizures in patients with Lennox-Gastaut syndrome and Dravet syndrome. Epidiolex is a non-psychoactive marijuana component that is currently being studied in phase III trials to treat epilepsy. Similarly, the phytocannabinoid cannabidivarin is also under study by GW Pharmaceuticals as a potential therapy for pharmacoresistant epilepsy.102, 108
Conclusion
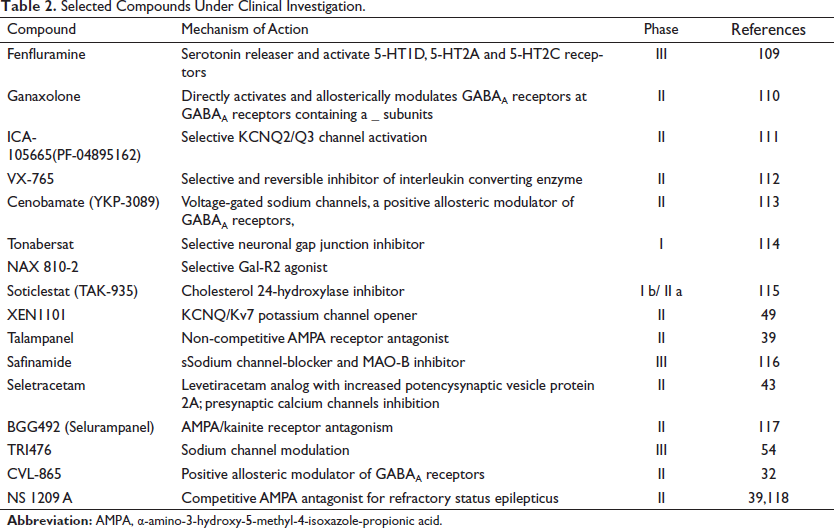
The currently available AEDs have several problems, including induction of congenital malformations, severe side effects, narrow therapeutic index, significant drug interaction, and lack of efficacy in several patients with epilepsy. Besides, AEDs act on symptomatic relief rather than solving the problems of epileptogenesis and pharmacoresistant epilepsy. Thus, several studies target several neglected signaling and pathophysiological pathways that contribute to the development and progression of epilepsy. The combination of existing AEDs with newer ones is under study for their synergistic effects. In this review, different signaling pathways are explained as promising targets to develop a new antiepileptic agent. Several preclinical studies revealed the development of agents that act on novel targets discussed in the review. Inhibitors including JNK, CLC- 2, GSK3β, mTOR pathway, CH24H inhibitor, and a monoclonal antibody for HMGB-1 showed great promising for disease modification and prevention. Table 2 contains some molecular agents that are in a clinical trial with a different mode of action. In the future, AED development should not only focus on disease-modifying agents but also prevent the onset and progression of epilepsy. However, drug development needs money, time, and new technology. In the future, we hope several AED will be approved for drug-resistant epilepsy.
Selected Compounds Under Clinical Investigation.
Footnotes
Abbreviations
Antiepileptic drugs (AEDs), central nervous system (CNS), voltage-activated calcium channels (VGCC), high voltage-activated (HVA), N-methyl-D-aspartate (NMDA), AMP (α-amino-3-hydroxy-5-methyl-4-isoxazole-propionate), Synaptic vesicle protein 2A (SV2A), Lactate dehydrogenase (LDH), Mitogen-activated protein kinases (MAPKs), c-Jun N-terminal kinases (JNKs), high mobility group box-1 (HMGB1), 24-hydroxylase (CH24H), Glycogen synthase kinase-3 beta (GSK3β), mammalian target of rapamycin (mTOR), G protein-coupled estrogen receptor 1 (GPER1), endocannabinoid system (ECS).
Acknowledgements
I would like to acknowledge Mrs Fasika Abu for editing the paper.
Authors’ Contributions
TM conceived the idea, drafted and revised the manuscript for intellectual content. The author read and approved the final manuscript.
Statement of Ethics
Not applicable
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship and/or publication of this article.