
Abstract
Background:
Spinal cord injuries incite varying degrees of symptoms in patients, ranging from weakness and incoordination to paralysis. Common amongst spinal cord injury (SCI) patients, neuropathic pain (NP) is a debilitating medical condition. Unfortunately, there remain many clinical impediments in treating NP because there is a lack of understanding regarding the mechanisms behind SCI-induced NP (SCINP). Given that more than 450,000 people in the United States alone suffer from SCI, it is unsatisfactory that current treatments yield poor results in alleviating and treating NP.
Summary:
In this review, we briefly discussed the models of SCINP along with the mechanisms of NP progression. Further, current treatment modalities are herein explored for SCINP involving pharmacological interventions targeting glia cells and astrocytes.
Key message:
The studies presented in this review provide insight for new directions regarding SCINP alleviation. Given the severity and incapacitating effects of SCINP, it is imperative to study the pathways involved and find new therapeutic targets in coordination with stem cell research, and to develop a new gold-standard in SCINP treatment.
Keywords
Introduction
The nature and extent of a spinal cord injury (SCI) is diverse and complicated. There are many symptoms, including, but not limited to, paralysis, myelopathy, and damage to white matter and grey matter. The complexity of injury is increased manifold as nerve fiber damage compromises sensation and motor signal transmittance to and from the brain, while grey matter damage results in segmental losses of interneurons. The utilization of corticosteroid (methylprednisolone sodium succinate), surgical interventions, and physiotherapy are the only methods for treatment in current health care, and these methods display limited success. 1 Yet, recent advances in the fields of stem cell biology have revolutionized neuroprotective and regenerative interventions.
Neuropathic pain (NP), because of its relatively unexplored molecular mechanism and widespread clinical morbidity, is extremely debilitating for SCI patients. In addition, NP is extremely resistant to treatment with current analgesic drugs, solidifying the necessity to find efficacious treatment options. Acknowledged in the current research are the emerging role of WNK1; cation-dependent chloride transporters (NKCC1) activation and inhibition by bumetanide2, 3; cannabinoid receptor (CB2) and anti-hyperalgesia effect of WIN 55,212-2 4 ; bradykinin (B1) and vanilloid-1 (TRPV1) receptor antagonists5, 6; and PPAR-gamma agonists in preventing neuropathic pain. 7 Time-specific changes in expression of matrix metalloproteinase-2 (MMP-2) in SCI-induced NP (SCINP) 8 and improved functional recovery with folic acid therapy has been found. 9 Likewise, bone marrow stromal cells (BMSCs) following lumbar puncture have shown some promising results in alleviating NP, including allodynia and hyperalgesia in chronic constriction injury (CCI) and spared nerve injury mice models. 10
Delving further into NP research, glia-mediated inflammatory reactions have been found to play a pivotal role in the introduction and development of NP. Microglia plays a fundamental role in proliferation, differentiation, and synaptic hemichannel growth in neurons. They are also known to be involved in the regulation of infection in brain tissue through innate and adaptive immune responses and maintaining homeostasis, respectively. Because of its major role in the neuroinflammatory process for neurodegenerative diseases, the study and utilization of microglia awakened from its relative dormancy. 11 A noteworthy discussion on microglial cells history 12 reveals their origin, differentiation, homeostasis, and implication in health and disease. For example, microglia have been recognized to have a critical role in Alzheimer’s, Parkinson’s, and Adrenoleukodystrophy. 13
Another glial cell type involved in providing neuroprotection is spinal cord astrocytes that release astrocytic mediators, for example, cytokines, chemokines, and growth factors for this purpose. 14 Unfortunately, the mechanism behind how astrocytes release astrocytic mediators is unclear, due in part to the lack of research on astrocytes because of their complexity in differentiation and seeding. Although astrocytic connexin-43 is implicated in gap junctions and communication of cytosolic contents via glial syncytia and to the extracellular space, the mechanism for this contribution remains unclear. Despite this, many studies have implicated astrocytes in facilitating or maintaining NP.
Studying molecular mechanisms, 15 discovered in the murine nerve injury model, MMPs activate and sustain NP. MMP-9 induces NP through interleukin-1beta cleavage and microglia activation at the acute stage. Similarly, latent stage MMP-2 maintained NP through the continuation of interleukin-1beta cleavage, though instead activating astrocytes. 16 Tissue inhibitors of MMPs (TIMPs) inhibit the activity of MMPs by regulating tissue proteolysis. As discussed, earlier, microglia and astrocytes help in tissue repair and breakdown in CNS. Therefore, studying the role of these glial cells in relation to NP may provide new insights into NP treatments. 17
Role of Glial Cells in Neuropathic Pain
Earlier pain induced after SCI was thought to be a result of anatomical, neurochemical, and excitotoxic alterations with changes in ion channels. 18 19 20 21 22 23 However, the current treatment modalities targeting neuronal activity by modifying the ion channels proved to be inadequate. 24 The focus was then shifted to neuron dysfunction, which until recently was thought to arise from neuroimmune modulation. Recent studies have made it evident that NP development involves complex mechanisms involving not only neuronal cells but also glial cells. These modulations are mainly contributed by the resident glial cells of the spinal cord. Astroglia and microglia are main cells attributing to inflammatory response and have been implicated in pain induction after injury. 25 SCI is followed by extensive neuroinflammation because of activation of these glial cells releasing chemokines and cytokines. 26 Thus, targeting neuroinflammation may open new treatment avenues for management of NP. 27
Implication of Microglia in Spinal Cord Injury-Induced Neuropathic Pain
Microglia play an important role in the maintenance and health of the CNS and are known to aide in neuronal differentiation and in the production of synaptic bonds. Microglia develop early in the embryonic yolk sac, contribute to brain development, and continue their work much into adulthood. The role of microglia in peripheral pain is well studied, and activation of these cells after partial sciatic nerve ligation, 28 spinal nerve ligation, 29 and sciatic nerve inflammation 30 have been found. Fractalkine, a microglial activator, induces allodynia and hyperalgesia shown by behavioral parameters. 31 Like peripheral injury, microglial activation has been reported in SCI 32333435, 23 and showed that the microglia shift from resting to an activated state in rats undergone T9 spinal cord contusion injury. These activated microglia contribute to chronic pain induction and maintenance after SCI. Microglial response at different timepoints determined that activation plateaued between two and four weeks after injury. In spinal nerve injury models, hyperactive microglia were found to increase the levels of P2X4 receptors. The expression was specific to microglia cells, while neuron and astrocytes remained unaffected. 36 P2X4 are purinergic receptors, and ATP is a known mediator of NP; therefore, it was suggested that upregulation of these receptors is linked to NP induction. However, the mechanism was not fully understood. Later, it was demonstrated that these receptors on stimulation lead to Brain-derived Neurotropic Factor (BDNF) secretion from activated microglia. 37 It was further found to affect NMDA receptors’ NR1 subunit in spinal cord dorsal horn neurons, which results in pain. This was supported by the studies in P2X4-deficient mice that, after peripheral nerve injury induction, display impaired BDNF signaling and lack hyperalgesia. 38 Another molecule, CCL21, was shown to rapidly express in injured sensory neurons. Further investigations in CCL21-deficit mice established the link between CCL21 and microglial P2X4. It was found that these deficit mice lack allodynia and reported with lower P2X4 expression. 39 Later it was demonstrated that a wide range of purinergic receptors were activated by ATP in response to nerve injury that led to microglial activation and subsequently NP (Multiple PY2). Molecular mechanism, such as those involving MAPK, ERK, and p38, have been implicated in signal transduction from microglial activation. For example, ROCK activation in spinal microglia has been shown with p38 MAPK activation and induction of NP. 40 Thus, it can be suggested that spinal microglia play a role in NP induction and may be explored for their use as potential targets for chronic pain treatment.
Long-term pain sensation is maintained by neuronal-glia interactions. Early phase and chronic phase are separately maintained by different mechanisms. Mice with nerve injury had persistent microglia activation for more than three months in the spinal cord. It was seen that microglia involvement is far beyond the cytokine and chemokine signaling that last for a limited period. Chronic inflammation by microglia, when targeted with immunotoxin Mac1-saporin, helps in pain reversal. 41 Evidently, microglia are strongly tied to inducing NP through MMP-9 activation in the spinal cord. The activation and deactivation of microglia through MMP-9 injection and inhibition has been demonstrated. 15 As an example, NAC attenuated remifentanil-induced postoperative hyperalgesia via inhibiting the cleavage of IL-1β, a substrate of MMP-9 in DRG, significantly inhibiting glial activation and neuron excitability in the spinal dorsal horn. 42
Implication of Astrocytes in Spinal Cord Injury-Induced Neuropathic Pain
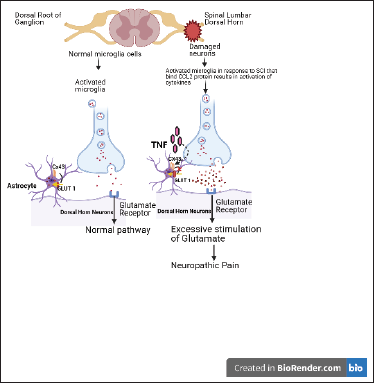
Astrocytes are cells lining the neurons and are involved in neuroinflammation by activating astrogliosis. Like microglia, long-lasting changes in astrocytes have been observed in in-vivo models of SCI. Astrogliosis is associated with development and NP persistence. Various mechanisms have been proposed through which astroglia contribute in NP. Astrocyte-related markers such as glial fibrillary acidic protein (GFAP) and aquaporin 4 are elevated in SCI rats. 34 Upregulated levels of GFAP and -p38 MAPK were also reported below and at level of injury, further supporting astrogliosis role in pain (Carlton et al. 2009). 43 Elevation of Connexin-43 (CX-43), a gap junction protein, points toward increased connectivity between adjacent astrocytes. 44 Wang and Xu 45 discussed the role and association of Cx-43 and pannexin channels in SCINP, and they concluded that CX-43 is an emerging therapeutic target for NP. Studies have also shown that these effects are a result of upregulation of Tropomyosin-related kinase B.T1 (TrkB.T1)-driven astrocytes (Figure 1). TrkB.T1 is an isoform of TrkB receptors that are expressed on astrocytes and an increased number of TrkB.T1+ cells after injury were reported that sustained through eight weeks. 46 In concordance with this, TrkB.T1 deletion in astrocytes led to reduced astrocyte proliferation in thoracic contusion SCI and improved hind limb withdrawal threshold. 47 Role of chemokines in activation of astrocytes has also been implicated in establishment of NP. Animals with spinal nerve ligation (SNL) have elevated levels of chemokines. CXCL13 has been shown to activate spinal astrocyte via another chemokine CXCR5. CXCR5-/-mice demonstrated lack of NP following SNL as CXCR5 was essential for activation of glial cells. Thus, neuron-astrocyte interaction lead to CXCL13 production by neuron cells that further activates astrocytes through CXCR5, inducing NP. 48 c-Jun N-terminal kinase (JNK)/monocyte chemoattractant protein-1 (MCP-1) pathway in astrocytes is also found to involved in NP development. 49 A separate study 50 bridges the role of both JNK1/2 and MMPs in NP where inhibition of astrocyte activation in the spinal cord by tetramethylpyrazine (TMP) prevented CCI-induced neuroinflammation. These findings demonstrate implication of JNK-MMP-2/9 in attenuating NP. In a mouse CPIP model, phosphorylated c-jun N-terminal kinase 1/2 (pJNK1/2) is downstream of spinal MMP-2. The MMP-2 inhibitor reversed the increase of glial fibrillary acidic protein (GFAP), the astrocyte biomarker, and pJNK1/2 on day three post injury. 51 The findings in this study include (a) increase in GFAP, but no significant effect on ionized calcium binding adaptor molecule 1, IBA1, a reactive microglial biomarker; (b) inhibition of astrocytes with fluorocitrate, but no inhibition of microglia with minocycline results in attenuation of allodynia in injured mice correlated with enhanced spinal levels pJNK1/2; (c) pJNK1/2 inhibitor, SP600125, showed decline in allodynia in injured mice; (d) increased expression levels of spinal MMP2, mainly NeuN a neuron biomarker; and (e) intrathecal administration of MMP-2 inhibitor, APR 100, resulting in delayed allodynia and decreased spinal levels of GFAP and pJNK1/2. Recent studies have also indicated that neuroinflammation plays a vital role in the occurrence and promotion of NP and that anti-inflammatory therapy has the potential to relieve the pain. 14 Thus, both microglia and astrocytes are linked to NP induction. In the next section, we will review studies that targeted these cells to ameliorate NP.
Glial Cells as Therapeutic Targets
Microglial Targets for Alleviating NP
Regulating microglia activity is thought to be a possible approach in impeding chronic pain progression. As discussed, activated microglia through production of various immuno-inflammatory molecules contributes to a state of chronic pain. These molecules lead to the activation of intracellular cascades in microglia cells generating and sustaining chronic pain. 11 Earlier studies with the goal of managing pain have explored the neuropathic roles of microglia and pharmacological interventions targeting activation of these cells.
Inhibiting p38 MAPK
Decades earlier, the glia cell-modifying functions of drugs like fluorocitrate and propentofylline had been shown to reduce pain sensitivity.52, 53 These drugs however did not distinguish glial cell types and which cells are responsible for pain sensitivity. Mounting evidence demonstrates the activation of microglia cells as exhibited by elevated levels of markers such as CD 11b and Iba 1. Phosphorylated p38 MAP kinase expression was also another characteristic feature of SCI and is believed to be another key regulator of NP. It was reported that p38MAPK was activated only in microglia after SCI. With the activation of p38, microglia produce proNGF via the p38MAPK-mediated pathway. When Minocycline was administered, it showed significant reduction in proNGF levels. The reduction was mediated by the inhibition of the p38 MAPK phosphorylation by the drug. Maintaining its levels using inhibitors has been shown to ameliorate NP. 54 Inhibitors of p38 have shown remarkable efficacy in reducing pain. SB203580, p38 inhibitor, has shown promising results in SNL-induced allodynia29, 36 while FR167653 or CNI-1493 has been reported to prevent NP in different neuropathy modes.55, 31 FR167653, another inhibitor of p38, reverses allodynia in SNL model for almost 6 h after single injection of 50 mg/kg, i.p. (Intra-peritoneal). Multiple injections of FR167653 maintained pain reception for two days after the last injection. Propentofylline given in nerve injury showed NP reversal by microglial response inhibition. 56 Likewise, efficacy of p38 inhibitor, SB203580 given intrathecally was seen only when provided before seven days of injury. 57
Altering Expression of Microglia Through Purinergic Receptors
ATP modulates microglial activity and is a ligand of the P2-purinoceptor family. Various P2 receptor subtypes such as P2X4, P2X7, and P2Y12 are expressed on microglia and are known to play a role in NP development. It has been shown that P2Y12, metabotropic purinergic receptor is linked to activated microglia and decreased expression of P2Y12, decreasing progression of NP. 58 Administration of P2X1-4 receptor antagonist, 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate (TNP-ATP) reverses tactile allodynia, while pyridoxal phosphate6-azophenyl-2′,4′-disulphonoic acid (PPAD), which inhibits P2X1–3, 5, 7 receptors without affecting P2X4 receptors, does not respond to tactile allodynia. This suggests that P2X4 receptor activation is required in pain stimulation. Antisense oligonucleotide against P2X4 receptors showed attenuated pain in nerve injury (Tsuda et al., 2003). Similar results were seen in genetic models with deleted P2rx4 gene. 38 These receptors are believed to stimulate pain induction through calcium influx and BDNF release 59 , further supported by experiments on P2X4 receptor-deficient mice showing impaired microglial BDNF expression. 38 Furthermore, mice deficient in P2X4 receptor that had undergone peripheral nerve injury showed no sign of mechanical allodynia strengthening BDNF and P2X4 association with NP. 60 61 62
Pain sensitivity reduction in P2X7 receptor-deficient mice indicates a role of these receptors in NP. 63 Pharmacological blockade using N-(adamantan-1-ylmethyl)-5-[(3R-amino-pyrrolidin-1-yl) methyl]-2-chloro-benzamide 64 or A-839977 65 has shown promising results. Likewise, Perez-Medrano et al., 2009 found positive effects of various cyanoguanidine antagonists of receptor for pain reduction. P2X7 receptor effects are mediated through the convergence to p38 MAPK signaling. However, while in P2X4 receptor-signaling, BDNF is a key molecule, P2X7 receptor signaling is regulated by the release of interleukin-1β and cathepsin S from microglia. 66 Apart from P2X receptors, microglia are known to have a wide range of P2Y receptors such as P2Y1, P2Y2, P2Y4, P2Y6, and P2Y12. 67 68 69 70 P2Y12 receptor is involved in eliciting pain sensitization. 57 Administration of 2Me-SADP, P2Y12 receptor agonist, mimics similar pain behavior in rats as nerve-injured animals. 58 Moreover, genetic manipulation of P2ry12 gene or suppression of expression through antisense oligonucleotides prevents mechanical allodynia.58, 71 Thus, it can be implicated that purinergic receptor mediated pain induction is an upstream process that converges to p38 MAPK signaling cascade.
Inhibition of Expression of Matrix Metalloproteinase/Induction of TIMPs Through Microglia
In peripheral nerve injury, Chattopadhyay 72 reported a dramatic increase of MMP-9. It was found that this upregulation of MMP-9 was linked with proinflammatory cytokines: TNF-α and IL-1β. Deletion of MMP-9 gene leads to significant reduction in NP, which further suggests the role of MMP in pain induction. 72 This opens a new avenue for NP management by MMP targeting. Mice with chronic constrictive injury (CCI)-induced NP, when treated with N-acetyl-cysteine (NAC), showed reduction in NP via a mechanism of MMP inhibition. 16 Both in vitro and in vivo experiments showed suppression of the activity of MMP9 and MMP2. NAC blocked the maturation of interleukin-1β, a substrate of MMPs, inhibiting the phosphorylation of protein kinase Cγ, NMDAR1, and mitogen-activated protein kinases. In this mechanism, NAC inhibited microglia activation but with no effect on astrocytes, thus demonstrating a safe and effective approach via strong inhibition of MMPS. TIMPs are endogenous inhibitors of MMPs 73 comprising of four inhibitors: TIMP1, TIMP2, TIMP3, and TIMP4. 74 Of these, TIMP1 and TIMP2 have specifically reported to alleviate the pain behavior by inhibiting MMP 2 and MMP 9.15, 75 Evidence of other small molecule inhibitors of MMPs that are employed to reduce the NP are reviewed. 76
Other Microglial Targets in Alleviation of NP
Although proliferation of microglia is correlated with NP, candidate molecules for this activation of spinal microglia remain elusive. A recent study reported that peripheral nerve injury induces microglial proliferation and pain through de novo expression of colony-stimulating factor 1 (CSF1) sensory neurons. 77 It was discovered that CSF1 binds to CSF1 receptors in microglia, which then activates and increases proliferation of microglia. Furthermore, they found that microglial membrane adapter protein, DAP12, is downstream of CSF1, which induces pain. 76 Likewise, recombinant Macrophage-CSF injection in rats induced microglial proliferation and development of mechanical allodynia, suggesting the role of M-CSF as a candidate molecule for induction of microglia proliferation. 78 Furthermore, Platelet-activating factor (PAF)/PAFr signaling has been implicated in peripheral nerve injury in spinal cord signaling. Autocrine or paracrine effects of PAF among the activated microglia and neurons have been shown in NP induction. 79 A recent study demonstrated that partial sciatic nerve ligation-induced pain can be attenuated by deficiency of lysophosphatidyl choline acyl transferase 2 (LPCAT2), a PAF biosynthetic enzyme. 80 Explored mechanisms exhibited LPCAT2 in wild-type spinal cord microglia, with no expression of LPCAT2 in LPCAT2-KO mice reduced spinal PAF expression. Also, pretreatment with PAF receptor antagonist ABT-491 showed a decline in ATP-stimulated PAF biosynthesis in macrophages designated as PAF–pain loop, thus demonstrating a novel therapeutic target that may lead to alleviation of NP. 80
Astrocytes Targets for Alleviating NP
Microglia have long been implicated in NP, unlike astrocyte involvement, which is still a new concept. Astrocyte activation is dependent on interaction between these cells and neurons, as well as release of factors from both cell types. In CPIP animal model, fluorocitrate inhibited activation of astrocyte in mice and attenuated the development of allodynia in them, while minocycline (microglia inhibitor) could not show the same effects. Thus, drugs targeting astrocytes are being explored for NP regulation rather than microglial pathway. 51
Astrocytic Glutamine-Signaling
Glutamate transporters are involved in pathological pain induction, and spinal astrocyte glutamate transporters are downregulated in pain. This aspect is also explored for possible therapy in chronic pain. In vivo imaging in rodent models has reported the role of metabotropic glutamate receptor five (mGluR5) signaling in S1 astroglia (Figure 1). Activation of this pathway induces allodynia, which is reversed by blocking the signaling. 81 Adenoviral infusion of GLT1 gene results in overexpression in astrocytes. Spinal GLT-1 gene transfer prevented the induction of partial sciatic nerve ligation–induced allodynia. 69 Consistent with these findings, riluzole inhibited NP by reducing extracellular glutamate by EAATs. 82 Moreover, ceftriaxone also prevented allodynia by selectively upregulating GLT-1 expression. 83 Taken together, it can be implicated that decreased astrocytic excitatory amino acid transporter in the spinal cord led to activation of glutamatergic synaptic pathway related to pathological pain. Propentofylline increases GLT-1 and GLAST expression and suppresses pain symptoms. 84 The roles of GLT-1 and GLAST are further supported by the use of amitriptyline, a first-line drug for the NP treatment, which is shown to reverse the GLT-1 and GLAST downregulation. 85
Blocking JNK Signaling in Astrocytes:
The studies related to translocator protein (TSPO) linked the activation of JNK signaling with chronic pain. Mouse model of spinal nerve ligation showed that Ro5-4864, TSPO agonist, reported reduced NP and that this was attributed to inhibition of astrocyte and p-JNK1 activation. Along with this, p-ERK was also reduced, suggesting the involvement of both JNK and ERK signaling. 86 Another JNK inhibitor, D-JNKI-1, ameliorated NP in SNL model of induced allodynia. 87 Later, MCP-1 was identified as main downstream molecule in JNK induced pain sensitization. 88 The association of JNK/MCP pathway was evident with the usage of drugs such as Tanshinone IIA (TIIAS) that targets this signaling and showed protection against NPin SNL animal models. Animals treated with TIIAS had elevated paw withdrawal threshold and reduction in astrocytic activation. Pathway analysis reported reduced JNK phosphorylation and MCP1 release in treated mice. 49 Fluorocitrate, an astrocyte inhibitor, provided protection in development of allodynia in CPIP-injured mice. This effect was suggested to be achieved through increased spinal levels of p-JNK. 51 SP600125 (JNK inhibitor) prevents the development of allodynia in the same mice model, and double immuno- staining showed colocalization of pJNK1/2 with GFAP, suggesting astrocytic involvement. Further studies revealed the involvement of matrix metalloproteinase-2 (MMP2) as they were upregulated. Intrathecal APR 100 (MMP-2 inhibitor) showed delayed development of allodynia with decreased levels of GFAP and pJNK1/2. This suggests the crosstalk between MMP2/JNK1/2 and MCP in astrocyte activation and pathogenesis of pain hypersensitivity. 51 Effectiveness of SP600125 in prevention of SNI and ddC-induced nociceptive behavior was also shown in another study where amitriptyline alone could not attenuate pain; however, when SP600125 was co-administered with amitriptyline, an antinociceptive effect was reported. 89 It was found that p-JNK was upregulated in SNI and ddC-exposed mice and that amitriptyline treatment further increased its expression. Additionally, it promoted astrocyte activation reported by elevated glial fibrillary acidic protein (GFAP) levels. Both JNK and glial activation was attenuated by co-administration of JNK inhibitor. Thus, it can be suggested that inhibiting astrocyte JNK activation exacerbates the amitriptyline analgesic response. 89 Modulating spinal astrocytes by targeting the JNK/MCP1 pathway can be an efficient approach against NP.
Other Astrocytes Targets for NP
Besides JNK and glutamate inhibitors, various other compounds have been tested for regulating astrogliosis in NP. One such compound is a well-known antioxidant, lycopene. Analgesic effects of lycopene were exhibited through prevention of Cx-43 protein downregulation. 90 Lycopene treatment increased the expression of Cx-43 protein in spinal astrocyte cultures in a TNF-dependent manner, which was found to be reduced in the NP model. Similarly, when mice with a partial sciatic nerve ligation were treated with repeated doses of lycopene, it resulted in inhibition of down regulation of Cx-43 expression in the spinal cord dorsal horn (SCDH) along with reduction in mechanical hypersensitivity. 90 Pioglitazone, a PPARˠ agonist, was shown to impart analgesic effects by reducing astrocyte activation, while administration of PPARˠ antagonist GW9662 abolished these effects, suggesting involvement of PPARˠ mechanisms. 91 Similarly, the analgesic activity of docosahexaenoic acid (DHA, 22:6 n-3) was confirmed in a rat model of a chronic constriction injury (CCI) where treatment with DHA showed reduced GFAP-positive astrocyte in the SCDH and ameliorated NP. 92
Future Directions
Although contemporary methods do not yield many results in the treatment of SCINP, glial cell-based therapies in SCI bring hope for the alleviation of symptoms and the development of a cure for NP. Because of the expansive research conducted for SCINP, there are many pathways that are available for targeting by current FDA-approved drugs and by other drugs that may still be in development. In a review, 93 it has been discussed that MMPs and TIMPs should be the primary focus of clinical studies because of their extensive studies and high implication of their function in NP. In addition to MMP-2/9, JNK-1/2, astrocytes, and microglia have been found to have a critical role in NP. Many studies have shown the specific roles these tissue types play in NP, and thus it is paramount to create a treatment of NP through targeting astrocytes and microglia. Studies have shown the effects of targeting each cell type independently, but there were no studies that demonstrated the treatment of both types of cells simultaneously. This raises the possibility of a dual treatment resulting in an additive effect, thus increasing the effectiveness and efficiency of the elimination of NP. Further studies should assess the effectiveness of a dual approach of treatment of astrocytes and microglia. Coupled with that, there are efficient methods to produce specific cell types such as astrocytes and microglia from stem cells to be utilized in both study and treatment. Thus, it may be time for this method to be utilized in a clinical setting as relevant human in-vitro studies and animal model have demonstrated exceptional promise. Furthermore, because of the lack of clinical relevance of translational animal models, it is advised that labs should consider the exploration of other models, such as the WMSTM model–relevant as a model for human spinal cord pathology and practical use as a platform for developing therapeutic delivery technologies. 94
Footnotes
Abbreviations
CX43, connexin; TNF, Tumor Necrosis Factor; GLUT1, Glutamate receptor 1.
NAC, N-Acetyl-Cysteine;
DRG, Dorsal Root Ganglia;
FDA, Food and Drug Administrations;
CXCL, C-X-C Motif Chemokine 13;
CXCR, C-X-C Chemokine Receptor;
NMDAR1, N-Methyl-D-Aspartate Receptors 1;
CPIP, Chronic Post-Ischemia Pain;
GLAST, Glutamate–Aspartate Transporter;
CNS, Central Nervous System;
ATP, Adenosine Triphosphate
NMDA, N-methyl D-aspartate;
NR, Hetero-Oligomeric Proteins Receptor Subunits;
CCL, Chemokine (C-C Motif) Ligand;
MAPK, Mitogen-Activated Protein Kinases;
ERK, Extracellular-Signal-Regulated Kinase;
ROCK, Rho-Associated Protein Kinase
Acknowledgements
We acknowledge Dr Robert J Dempsey, Chairman, Neurological Surgery, for his constant encouragement during the study.
Authors’ Contribution
All the authors contributed to concepts, definition of intellectual content, literature search, manuscript editing, and review.
Statement of Ethics
The paper reflects the authors’ analysis in a truthful and complete manner. The paper properly credits the meaningful contributions of co-authors and co-researchers. This material is the authors’ own original work. The paper has not been previously published. All sources used are properly credited.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.