
Abstract
Objectives
To evaluate the French cystic fibrosis newborn screening algorithm, based on data tracked by a centralized monitoring process, from 2002 to 2014. The programme aimed to attain European Standards in terms of positive predictive value, sensitivity, the ratio of screen positive patients diagnosed with cystic fibrosis to infants who screen positive but with inconclusive diagnosis (CFSPID), and time to diagnosis.
Methods
Retrospective analysis of programme performance, compliance with the algorithm, and changes in screening strategy.
Results
Modifications in the flow chart protocol improved the positive predictive value to 0.31 while maintaining the sensitivity at 0.95. Among infants diagnosed with cystic fibrosis, or identified as CFSPID, sweat test results were obtained for 94%, and two mutations were identified after exhaustive screening for the gene, when applicable, in 99.6%. The rate of pending diagnosis was very low (0.5%). The ratio of infants with cystic fibrosis:CFSPID was 6.3:1. Age at initial visit at the CF centre was ≤ 35 days, respectively, in 53%/26%.
Conclusion
Performances were in agreement with European standards, but timeliness of initial visit needed improvement. Our data complement an accumulating body of evidence demonstrating that attention must be paid to such ethical considerations as limiting carrier detection and inconclusive diagnosis. Newborn screening programmes should have a rigorous centralized monitoring process to warrant adjustments for improving performance to attain consensus guidelines.
Keywords
Introduction
Newborn screening (NBS) offers the opportunity for early diagnosis and improved outcome in patients with cystic fibrosis (CF).1–3 It is performed worldwide but with considerable variability in strategies.4,5 Important drawbacks include false-positive test results, detection of carriers, infants who screen positive but with inconclusive diagnosis (CFSPID), and missed CF cases. In the French newborn CF screening programme, a centralized monitoring process was implemented by the Association Française pour le Dépistage et la Prévention des Handicaps de l’Enfant (AFDPHE) to collect data from screening laboratory centres (22 laboratories for immunoreactive trypsinogen (IRT) measurements, 9 for mutational analysis, 22 regional NBS centres, and 37 CF centres) and to optimize screening strategies with adjustments to biological cut-off levels, mutation panels, or changes in the algorithm. The objective was to attain European Cystic Fibrosis Society best practice standards of care 6 in terms of positive predictive value (PPV), sensitivity, ratio of patients diagnosed CF:CFSPID, and time to diagnosis. We analysed and summarized a retrospective evaluation of the national programme since 2002.
Methods
Screening algorithm
The CF-NBS programme employed a three-tiered algorithm with an initial IRT-1 assay (Delfia Neonatal IRT kit – Perkin Elmer) at day 3, followed by a CFTR panel mutation testing (CF20 Elucigene® kit then CF30 from 2004) when the IRT result was above the cut-off and if written informed consent was obtained on the back of the Guthrie card. (Note: CF30 Elucigene® kit (legacy mutation nomenclature): F508del; I 507del; 1078delT; 1717-1 G > A; 2183AA > G; 3659delC; 3849 + 10kbC > T; 621 + 1G > T; A455E; E60X; G542X; G551D; N1303K; R1162X; R117H; R334W; R347P; R553X; S1251N; W1282X; 1811 + 1,6kbA > G; 2789 + 5G > A; 3120 + 1G > A; 3272-26A > G; 394 delTT; 711 + 1G > T; G85E; Y1092X; Y122X; W846X.) A second IRT was performed at day 21 (IRT-2), if there was no parental written consent or no identified mutation. In 2003 and 2004, analysis of data by the technical committee of the AFDPHE led to changes in screening strategy to optimize the programme (as previously published 7 ) by simultaneously increasing the IRT-1 cut-off (from 60 to 65 µg/L) to maintain the target of the 0.5% positive test, and the IRT-2 cut-off (from 30 to 40 µg/L) because of the very low rate of diagnosed CF in the absence of detected mutations. The IRT-2 sample was maintained in cases with no written consent for mutational analysis, but was restricted to newborns with no identified mutation and a very high IRT-1 (≥100 µg/L).
Definitions and terminology
Diagnosis
Diagnosis in newborns screen positive for CF was confirmed by a sweat chloride concentration of ≥60 mmol/L (some centres used conductivity, with cut-off values published by Nguyen et al. 8 ), or two CF causing CFTR mutations in trans, 9 with the exception of those carrying a c.3718-2477 C > T (3849 + 10kbC > T) mutation known to be associated with sweat test values below the cut-off.
CFSPID
This is a descriptive rather than a diagnostic label, as these infants do not have disease, but have a number of risk factors for developing CF-related conditions in the future. It is applied to those who screened positive with either a normal sweat chloride result and two CFTR gene mutations, at least one of which had unknown phenotypic consequence, or an intermediate sweat chloride result and one or no mutations. 10
Carriers
Newborns screened positive were labelled as carriers based on identification of one mutation in the CF30 kit and a normal sweat chloride concentration <30 mmol/L.
Pending status
This refers to screen positive infants referred at a CF centre where the CF physician cannot provide a definite conclusion, either because the sweat test or the allele mutational analysis is still ongoing.
PPV
This is the number of infants with a true positive NBS test divided by the total number of positive tests.
Sensitivity
This is the number of true positive NBS results as a percentage of the total CF population (true positive and false-negative); for the calculation we included in the analysis cases missed by NBS that resulted in delayed diagnosis (excluding meconium ileus (MI) or prenatal diagnosis (PND)).
Programme surveillance
For each screen positive infant requiring referral for sweat testing, a pre-filled document (including IRT results, molecular genetics, term pregnancy, birth weight and length) was faxed by the regional NBS centre to the nearest CF centre to the home address; receipt was confirmed by telephone. The CF physician called the family to arrange an appointment for a sweat test on the same or the following day, and then completed the form, noting date of initial visit, symptoms, sweat test results and outcome, and with a conclusion code corresponding to: CF (CF or CFSPID and including MI, PND, and familial history of CF); non-CF; pending status; death; or lost to follow-up). The form was returned to the regional NBS centre and forwarded quarterly, after partial de-identification, to the centralized NBS structure (AFDPHE), where a referent CF physician and national data manager were in charge of monitoring. CFSPID was identified centrally, case by case, based on CFTR mutations 9 and sweat test results. A board of physicians, a midwife, an ethicist, and biologists met monthly to discuss all NBS programmes, and every three months the technical committee reviewed monthly distribution of IRT results and molecular genetics, refining practical aspects of the CF NBS programme. Compliance with the algorithm, time to initial visit to the CF centre, sweat test results, and complete genotype in those diagnosed with CF or identified as CFSPID were tracked. Regular queries and updates of initial missing information were part of monitoring. In addition, an annual questionnaire was sent both to CF and regional NBS centres, to collect cases missed by NBS but diagnosed based on symptoms. Combined analysis of laboratory and clinical outcome data, so as not to increase the rate of missed cases, led to changes in IRT cut-off levels and screening strategies for limiting the number of false-positive cases and detecting infants identified as CFSPID. The AFDPHE national database was recently linked to the French CF patient registry database 11 to provide the most comprehensive data.
Statistical analysis
We evaluated the characteristics of the screening protocol, mainly PPV, sensitivity, and incidence. Qualitative variables are described as numbers and percentages, and quantitative variables as medians [Q1–Q3]. Incidence rates are given with their 95% confidence interval (CI). For comparing groups, chi-square test was used for comparing medians, and the student t-test for percentages and other quantitative variables. Level of significance (p-value) was set at 5%. Statistical analyses were performed using SPSS Statistics v19.0 software.
Results
Study population
Data on the newborn CF screening population during 2002–2014 in accordance with changes in the algorithm and data on the false negative cases during 2002–2013.
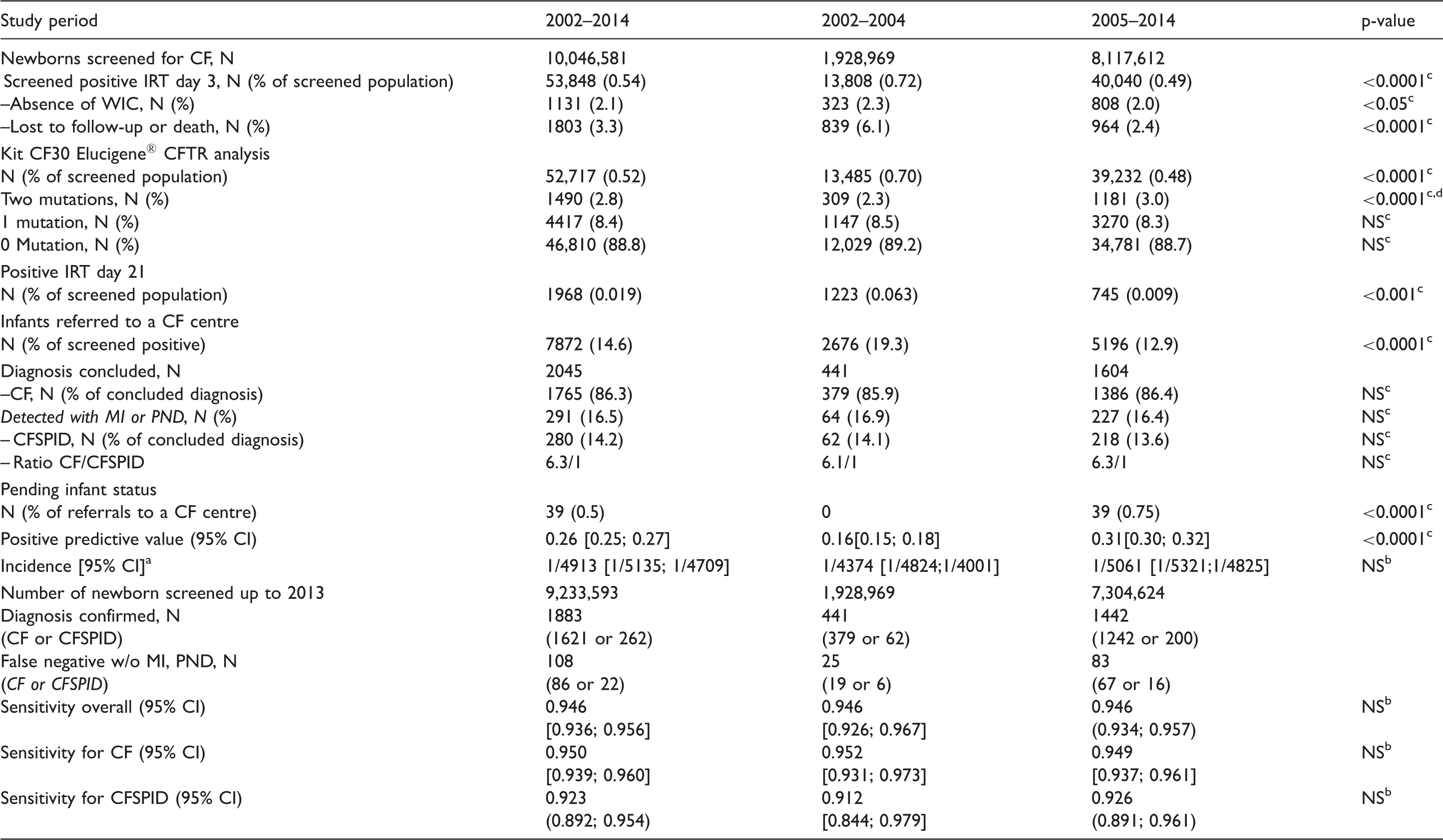
aCF and CFSPID. bChi square test. cStudent's t-test. dKit CF20 moved to CF 30. WIC: written inform consent; MI: meconium ileus; PND: prenatal diagnosis; CFSPID: CF Screen Positive, Inconclusive Diagnosis.
Reported sweat testing results and molecular genetics
The rate of reporting of sweat test results in infants diagnosed with CF or identified as CFPSID carrying two CF30 mutations, excluding MI or PND, was 1226/1284 (95.5%). In the cohort with MI or PND (228/291), it dropped to 78.3%. For infants carrying one mutation or with no mutations, and excluding cases of MI and PND, CF centres did not provide sweat test results despite reminders, for two out of 470 infants. Nevertheless, infant status was determined based on familial CF history and positive mutational analysis. The CF30 kit identified 87.7% of CF alleles, with a detection rate of ≥80% for all 22 French regions. Mutational analysis in the 2045 infants with CF or identified as CFSPID was 4073/4090 (99.6%), as only 17 alleles were unidentified after CFTR next generation sequencing methods, including the search for large rearrangements.
CFSPID and the p.Arg117His mutation
Characteristics of CF-screened positive cohort carrying at least one p.Arg117His (period 2002–2012, number of newborns screened: 8,420,082).
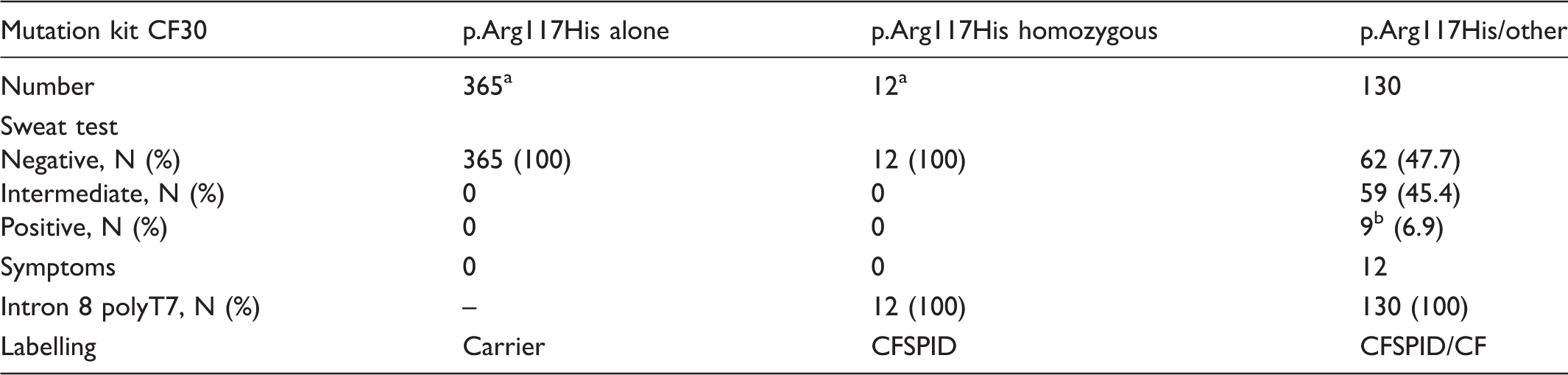
Exclusion of p.Arg117His would ignore them.
All these cases were symptomatic.
CFSPID: CF Screen Positive, Inconclusive Diagnosis.
Time to initial visit at the CF centre and symptoms
Age and symptoms at initial visit to a CF centre of infants diagnosed with CF and identified CFSPIDa (period 2002–2014, number of newborn screened: 10,046,581).
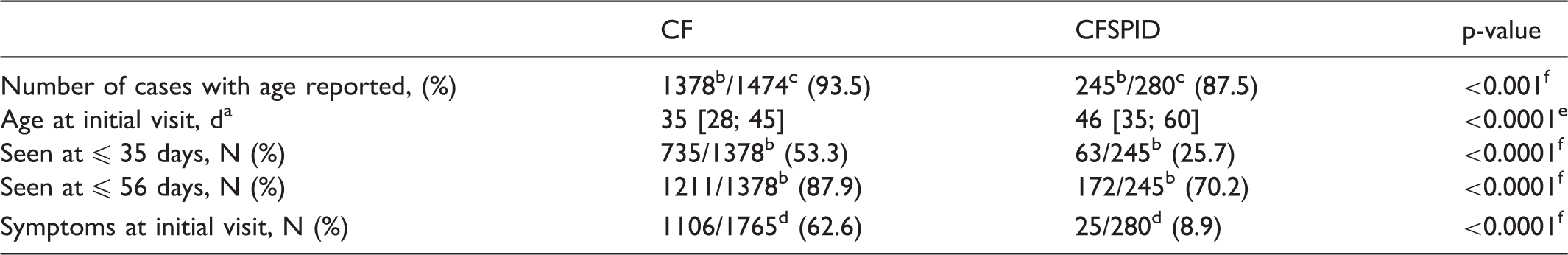
aMedian [Q1–Q3]. bAvailable data. cExcluding meconium ileus and prenatal diagnosis. dIncluding meconium ileus and prenatal diagnosis. eChi square test. fStudent's t-test. CF: cystic fibrosis; CFSPID: CF Screen Positive, Inconclusive Diagnosis.
Discussion
This study demonstrates that a rigorous surveillance tracking process can evaluate the performances of a national programme. Missing parental informed consent for DNA analysis was mainly related to newborn hospitalization rather than parental refusal, and all such infants followed the algorithm with IRT-2 sampling. Adjustments in the protocol achieved a PPV of 0.31 while maintaining 95% sensitivity, in agreement with the European Cystic Fibrosis Society Standards of Care. 6 Infants carrying one mutation in the CF30 panel were referred for a sweat test, with a diagnosis of CF or labelled CFSPID in 11% (Table 1), while the remaining infants were carriers. We had a very low rate of pending status; 54% were born in 2013–2014, and information on sweat testing and mutations is expected shortly.
We maintained the IRT-2 sampling for sensitivity purposes, because we identified 42 infants with CF (1/18) among the 745 with positive IRT-2, related to screened population diversity and limited CFTR panel mutations. In the literature, fail-safe protocols vary from a second IRT sampling to immediate sweat testing in cases of very high initial IRT or expanded mutation panels, such as in Massachusetts; however, low PPV at 0.09 12 is considered unacceptable. The UK programme is remarkable for its excellent PPV of 0.75 13 ; it combines a limited panel of four CFTR mutations, followed by a second step panel mutation (29/31 mutations) if only one mutation is detected; this avoids the need for sweat testing in cases with only one mutation after the second-step mutation panel with low IRT-2.
We found strong adherence to sweat testing (95.4%) in those carrying two CF-causing mutations. NBS is not a diagnostic test, and whether or not the infant has CF relies on demonstrating CFTR dysfunction; furthermore, sweat testing is a safeguard against mislabelling newborns, laboratory errors, or mutations carried in cis. 14 Improving adherence to sweat testing remains challenging in many European countries 15 and in the US, with nearly 24% of cases without sweat test results. 16
We report comprehensive identification of both alleles after complete CFTR analysis, 17 when applicable. This is in agreement with diagnostic criteria for CF 6 defined by sweat chloride above 59 mmol/L and/or two CF-causing CFTR mutations in trans. Mutation panel sizes vary from a few mutations to a comprehensive panel, as in California 18 or Poland, 19 thus identifying many more carriers and CFSPID; this is in disagreement with the European Cystic Fibrosis Society Standards of Care. 6
One of the most controversial negative impacts of genetic analysis in NBS algorithms is detection of babies carrying at least one non CF-causing mutation, as most of these infants will remain asymptomatic. The p.Arg117His mutation was overrepresented 17 consistently on a T7 background, considered neutral in light of the statement that p.Arg117His T7 should no longer be considered a CF-causing mutation in asymptomatic newborn infants. 20 The technical committee of the AFDPHE validated the consequences of p.Arg117His removal (see Table 2); CF centres then agreed to remove this mutation (61%) and Elucigene® confirmed the feasibility of CE-IVD certification. The new kit excluding p.Arg117His was introduced in 2015. Analysis of data collected in 2015 showed dramatic improvement in the ratio CF/CFSPID (9:1), a decrease in carriers, and increased PPV (0.34). Thus, in neonates who screened positive with one or no CFTR mutation and a sweat test ≥30 mmol/L, a search for other mutations was required, and p.Arg117His was included in this second-line panel. In the literature, very few case reports carrying a severe mutation and p.Arg117His T7 had early pulmonary manifestations.21,22 Removal of p.Arg117His cannot be extrapolated to countries in which the polypirimidine variant in intron 8 in cis is on a T5 background, due to its associated pathogenicity. Recently, Norway 23 introduced NBS for CF with a mutation panel immediately excluding p.Arg117His T7, and The Netherlands removed it in July 2016.
Median time to initial CF centre visit in those diagnosed with CF (excluding MI and AND) was 35 days, 24 except for 2013. These cases were individually analysed at each of the four main steps of the screening protocol: delay from IRT sampling to results (>10 days: n = 4), mailing of Guthrie cards to the genetic laboratory (>10 days: n = 3), mutational analysis (>15 days: n = 15), and initial visit to the CF centre (>15 days: n = 32). The AFDPHE board and the technical committee sent letters to all partners providing suggestions for improvement, such as sending Guthrie cards to genetic laboratories daily rather than weekly, immediate secure faxing of positive results of mutational analysis to the regional NBS centre, greater vigilance during holidays when laboratories and CF centres are understaffed, and request for an explanation if referral (defined as the date on which the infant was first seen, rather than the date of successful sweat testing) to a CF centre was delayed. In the following year, 2014, median delay to initial visit returned to a baseline of 35 (28; 41) days. However, 12% of infants with CF were seen beyond 56 days, and this remains a crucial point requiring improvement, as the rate of respiratory symptoms was higher in those seen after 35 days. The European Cystic Fibrosis Society Standards of Care 6 statement specifies that “the majority of infants with a confirmed diagnosis after NBS should be seen by the CF specialist team by 35 days and no later than 58 days after birth.”
Our study has some limitations. First, we could not provide a case-by-case explanation for non-written informed consent for DNA analysis, as this is not requested on the Guthrie card. In addition, the rate of initial sweat testing failure was not evaluated, and the results reported in the document might relate to older infants.
Our strategy was considered unethical by the National Ethical Committee (No. 57 01/2007), because detection of carriers via CFTR analysis was limited to families with an infant having high IRT-1 (1/2000 instead of 1/33 in the general population), but was considered beneficial by many parents in terms of reproductive planning. 25 Later, the Ministry of Health (HAS, 2009) concluded that the programme gave satisfactory performance, but confirmed that healthy carrier detection and CFSPID identification were beyond the goals of NBS and should be avoided by alternative strategies. It considered the panel of mutations based on frequencies in the general population to be unfair to ethnic minorities, and requested removal of DNA analysis. Thus, a study comparing the current strategy with IRT/pancreatitis-associated protein (PAP) protocol was conducted on 500,000 newborns in 2010. Data demonstrated non-inferiority in terms of sensitivity, no carrier detection, and a decreased rate of CFSPID identification, but a dramatic decrease in PPV (0.09), 26 that represented a major barrier to implementing this strategy. Recently, European experiments combining IRT/PAP/DNA27–29 showed similar sensitivities and much better PPV than IRT/PAP, but a residual number of detected carriers and CFSPID. Cost of IRT/PAP/DNA and IRT/DNA strategies was similar. Based on these data, an IRT/PAP/DNA strategy with a similar financial envelope to the current IRT/DNA was submitted to French authorities and to Health Insurance for agreement. Currently, the technical committee of AFDPHE is working on IRT and PAP cut-offs values, and on the usefulness of a safety net for evaluating the feasibility and financial cost.
In summary, despite its worldwide use for many years, NBS for CF presents a number of challenges, including finding a satisfactory balance between positive predictive value, sensitivity, and clinical utility.30,31 Evaluation of the performance of ongoing algorithms must rely on rigorous centralized tracking, aimed at monitoring outcome data. The newborn screening process begins at the maternity ward, but ends at the CF centre, with a definitive conclusion, enabling those diagnosed with CF or identified as CFSPID to rapidly benefit from adequate care management and follow-up. Our data complement an accumulating body of evidence demonstrating that attention must be paid to such ethical considerations as limiting carrier detection and inconclusive diagnosis.
Footnotes
Acknowledgements
We acknowledge all the maternity wards, screening laboratories, molecular genetics laboratories, regional NBS centres, and physicians at CF centres for their close collaboration in providing data.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.