
Abstract
Interpenetrating polymer networks of vinyl ester (VE) resin and polyurethane (PU) were synthesized using blend ratio of 93:7(w/w). Two sets of nanocomposites based on i) pure vinyl ester and ii)VE/PU IPN(93VE), were prepared with organically modified silica nanoparticle (OMS) as filler by 1, 2, 3 and 5% weight of the matrix resin. All the nanocomposites were characterized in terms of mechanical and thermomechanical properties.VE/silica nanocomposite with 2% filler (VES2) showed improvement in ultimate tensile strength by 83.5% and toughness by 42% compared to that of VE resin itself. The IPN based nanocomposite, 93VES2, exhibited 31.14%, 10.8% and 18%greater tensile strength, Young’s modulus and toughness respectively in comparison to that of the base 93VE IPN. IPN based nanocomposites were tougher than VE based nanocomposites. Storage modulus of nanocomposites was lower than that of 93VE and VE matrix system. Higher tanδmax of the 93VE/OMS nanocomposites than that of the 93VE matrix was indication of more elastic nature of the later. Smaller size of dispersed domains was found in SEM micrographs for IPN based nanocomposites than that in micrographs of VE based nanocomposites of corresponding composition.
Keywords
Introduction
Vinyl ester (VE) resin is widely used in coating, adhesives, and fiber-reinforced composite formulation owing to its high strength and modulus, low shrinkage, and excellent thermal stability. However the resin suffers from brittle behavior when it is cured. 1 The toughness of VE had been increased by blending with an elastomer, 2 a thermoplastic, 3 another thermoset 4 and by the incorporation of inorganic nanoparticles. 5 Blending two polymers, being an effective way to devise novel polymer materials with optimized properties, was tried in the present work for the improvement in toughness of VE.6,7
Formation of interpenetrating polymer networks with epoxy resin, polyurethane etc. to enhance the toughness, thermal stability and mechanical strength, are reported by Song et al. 8 Tang et al. 9 prepared a series of PU based gradient IPNs from Polyurethane/Vinyl Ester Resin IPN systems, which has the better mechanical and thermomechanical properties than conventional IPN.
Parmar et al. 10 reported that the three-component IPN matrix based glass fiber-reinforced composites made with the vinyl ester of an epoxy novolac resin, diglycidyl ether of bisphenol-A and MMA. They found that it is resistant to the chemicals and has better thermal, mechanical properties than neat resin.
Hui Jin et al. 11 prepared the IPN based on epoxy resin and polyurethane pre-polymers to improve the damping properties but has poor tensile modulus and strength than pure epoxy. It also shows the improved toughening of epoxy matrix.
Wang and Chen 12 modified diglycidyl ether of Bisphenol-A with a PU pre-polymer terminated with aromatic amine groups as well as phenolic hydroxyl groups. They showed that the epoxy modified with PU containing phenol hydroxyl groups had higher fracture toughness than the corresponding system. Hsieh and Han 11 shows that the significant improvement in the tensile strength of graft IPN of an epoxy and Polyurethane which was correlated to the grafted structure and to the length of PU chains.
Chen et al. 13 made polyurethane based IPNs on TDI and graft polyol and vinyl ester resin for reactive injection molding process and found that the IPN prepared with graft polyol has best mechanical properties among all specimens.
Nanoparticles provide large surface area which promotes the formation of primary valence bonds at the filler/resin interface and allows effective stress transfer across the interface. 14
The use of thermoset–thermoplastic blends or IPNs and nanocomposites, obtained by incorporation of inorganic nanoparticles have gained much attention nowadays due to their enhanced mechanical properties and thermal stability.15,16 Organically modified nanofiller was tried in many cases instead of normal nanofiller to enhance the resin/filler interaction further.
Therefore, in the present research, organically modified silica nanofiller is added to neat vinyl ester resin and VE/Polyurethane IPN to find out their performance in comparison to the VE resin alone. The characteristic features of VE lies in between that of epoxy and unsaturated polyester due to its unique chemical structure. PU is a class of very useful and versatile material and widely used for various applications.17,18 Its unique acceptance is due to the elasticity, toughness and durability in adverse pH conditions, temperature, humidity etc. 19
The present work comprises of synthesis of VE/PU based graft IPN in which VE is the major component. Use of VE as the major component in the IPN is aimed at the retention of high tensile strength and thermal stability of the thermoset resin, as required for structural components. In addition, the PU is used as crosslinks between the chains to improve the toughening of IPN. During IPN formation, the interaction between the component polymers is possible at molecular segment level because of simultaneous in situ formation of PU polymer and crosslinking of VE at respective pre-polymers. Hypothesis of the present research is based on the reaction of polar hydroxyl of VE with isocyanates of PU pre-polymers which may lead to compatibilization at the interface of the two components.20,21 Blend preparation is started with pre-polymer of PU and uncrosslinked VE premixed with styrene diluent. Crosslinking of VE is to occur through free radical polymerization involving C=C on the VE while grafting of PU chains on VE would occur through condensation reaction as was observed by others.
Unlike previously reported works here VE/PU graft IPNs are prepared with VE as major component. Secondly, organically modified silica nanofillers are incorporated in the IPNs as well as in virgin resin by similar concentration and comparison is done to show the improvement in properties of the composites due to IPN matrix. Optimization of blend ratio of the IPNs was done on the basis of the mechanical and thermomechanical properties of them. Using the IPN of optimum composition as matrix, nanocomposites were prepared further with organically modified nano-silica. The advantage of the whole process lies in that it was carried out at low temperature and nanocomposites of VE and VE/PU IPN resin were prepared by simple casting technique.
Experimental
Materials
Ruia Chemicals Pvt. Ltd. of New Delhi, supplied vinyl ester resin (tir @bond 701), which was used as the major component in the matrix. Properties of this virgin resin are given in Table 1.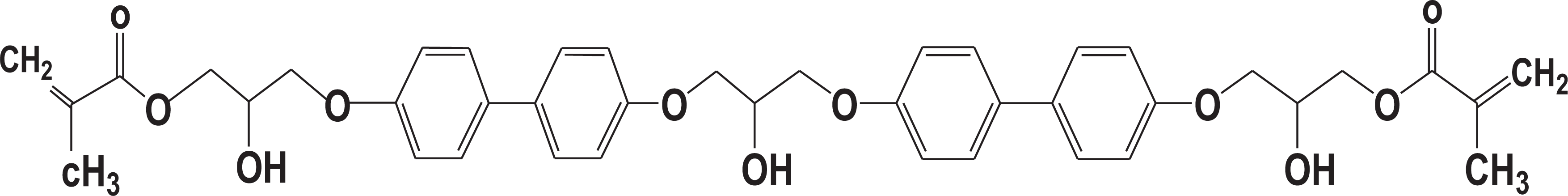
Properties of VE resin.

N, N’-Dimethyl aniline (DMA), Methyl ethyl ketone peroxide (MEKP) and Cobalt(II) naphthenate were used as a promoter, initiator and catalyst respectively for curing of VE resin. These were procured from the resin supplier. PU (Alprothane 4095), a liquid isocyanate terminated (4.6% NCO) pre-polymer based on polypropylene glycol, was procured from Lympro Chem Private Limited, Mumbai, India. It was used without hardener.
Organically modified Silica (OMS) i.e. silica coated with KH570, was purchased from Nanoshell (Haryana, India). Particle Size of the dark brown silica powder was 20–30 nm with spherical morphology.
Preparation of vinyl ester/polyurethane interpenetrating polymer networks (IPN)
Vinyl ester resin, premixed with 45% styrene as diluent, was further mixed with polyurethane pre-polymer in definite proportion (93:7 w/w), under constant stirring in a water bath sonicator for 30 minutes and was left at room temperature till the bubbles disappeared (Figure 1). Then MEKP, cobalt naphthenate, and DMA (2 wt. % each) were mixed uniformly with this resin mixture and kept at rest till the bubbles disappeared. Casting of the mixture was done by pouring into channels of dimension 120 × 15 × 3 mm3 in a PTFE mold and kept at room temperature for solidification. Finally, curing was carried out in an air oven at 40°C and post cure at 80°C in vacuum oven for 4 hours. Pure crosslinked vinyl ester sample was named as VE while the IPN was designated as 93VE.
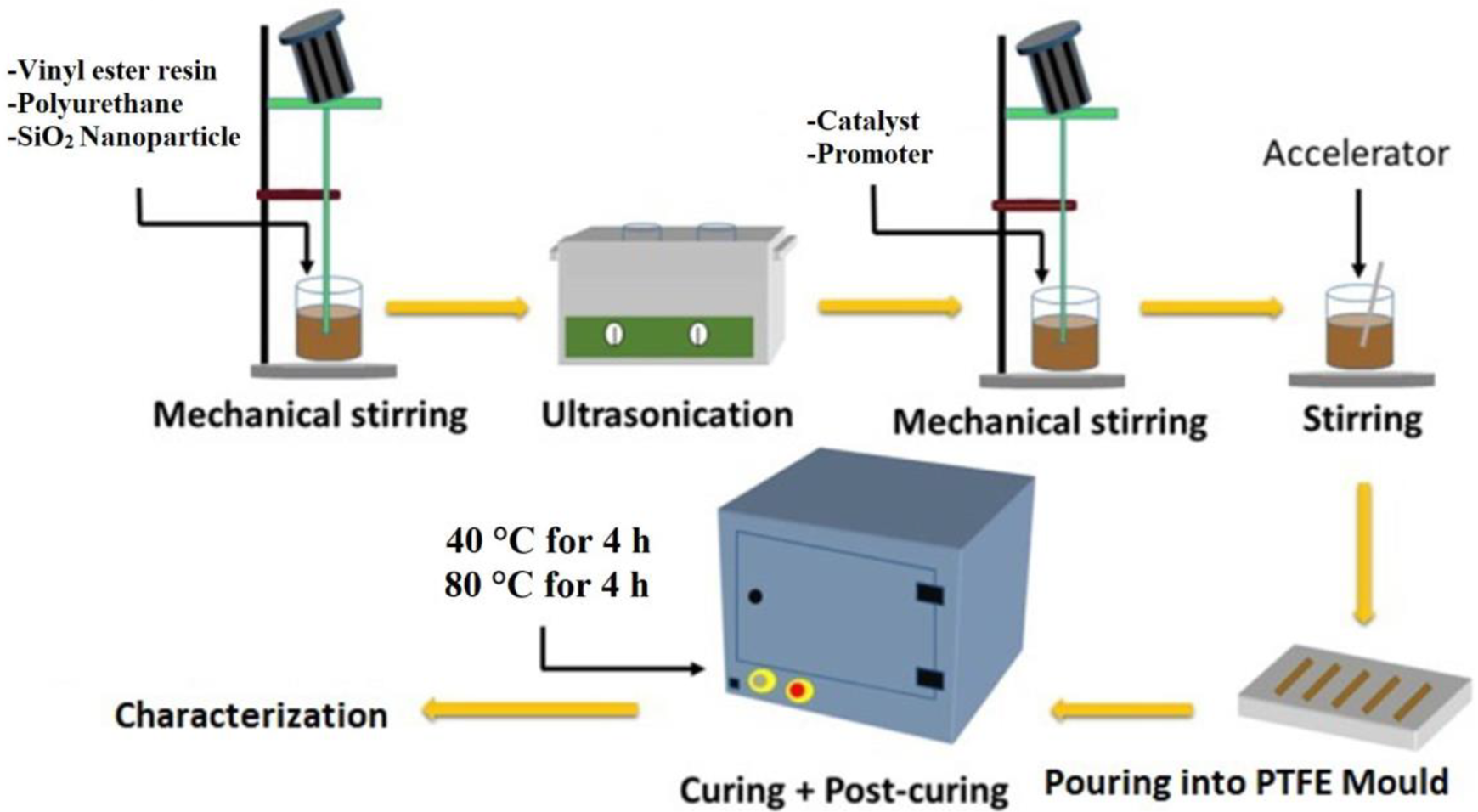
Preparation of VE/PU IPNs.
In the present study, VE was crosslinked in presence of linear PU pre-polymer chains. By definition of IPN one polymer (VE) was crosslinked while the linear chains of other polymer (PU) got entangled with the in situ formed network of VE. Also here intermolecular reaction involving –NCO of PU and –OH on VE chains is feasible. 20 Therefore PU chains may get grafted on the VE chains through –NHCOO linkages; hence it may be termed hereafter as “Graft Interpenetrating Network” or GIPN.
VE and PU resin mixture, mixed with 1, 2, 3 and 5 wt.% of silica nanoparticles, were prepared by the same procedure as shown in Figure 1 in which silica nanoparticles were mixed before ultra-sonication. The mixture was sonicated in water bath sonicator for 20 minutes at room temperature. In the next stage, catalyst, promoter and accelerator for vinyl ester curing were added by the same concentration as mentioned earlier to the resin system and mixed.
Vinyl ester nanocomposite samples were named as VES1, VES2, VES3 and VES5 in which the amount of OMS nanoparticle was 1%, 2%, 3% and 5% by weight respectively with respect to the total weight of liquid resin. OMS filled IPN nanocomposites were designated as 93VES1 etc. Where the first number stands for the weight ratio of VE to PU in the IPN i.e. 93:7(w/w) and the second number is for the weight of nano-silica (%) in the liquid resin taken for IPN synthesis.
Characterization
Morphology
Surface morphology of specimens was observed using scanning electron microscope Carl Zeiss Microscopy, Oxford Instruments Nano analysis, at an accelerating voltage of 20 KV and at 20KX magnification.
Fourier transform infrared spectroscopy (FTIR)
The FTIR analysis was carried by Shimadzu Corporation, Japan (Prestige-21). The powdered sample was prepared by crushing the solid sample and mixing with KBr and forms the pallets. The spectra were taken at a resolution of 4 cm−1in a scanning range of 400 to 4000 cm−1 in ATR mode.
Specific gravity
Specific gravity of crosslinked vinyl ester resin and IPNs were determined by using Archimedes principle as in equation 1

where A and B are weights of the sample in air and water, respectively and

where, g1 and g2 are specific gravity of polymer1 and polymer 2 components in IPNs. Weight fractions of polymer 1 and 2 are w 1 and w2 respectively. Equation (2) can be extended for IPN based nanocomposites to observe the effect of specific gravity (g3) and the weight fraction of filler (w 3 ) of the nanofiller as (Equation 3).

Crosslink density
Crosslink density of various resin systems was determined according to the classical Flory–Rehner method 22 as shown in Equation 4.

where ‘γ’ is crosslink density; Vp = volume fraction of polymer in the swollen mass; Vs is molar volume of the solvent; dr is density of the crosslinked resin; is polymer–solvent interaction parameter. The swelling of each sample was carried out at 27°C using nine different liquids ranging in their solubility parameter from 28.84 (MPa)1/2 to 51.55 (MPa)1/2. The percent increase in weight of each of these samples, after swelling in the different liquids, were noted in regular intervals of time till the equilibrium swelling was reached. The swelling coefficient Q was calculated using equation 5.

where, m and m0 are weight of the sample and dry sample respectively,

The polymer–solvent interaction parameter was then calculated from Bristow and Watson equation 23 as given in equation 7:

where, β = lattice constant = 0.34; R, universal gas constant (J/K/mol); T, absolute temperature (K); δ s and δ p are solubility parameters (MPa)1/2 of the solvent and the IPN sample, respectively.
Thermomechanical properties of VE and 93VE IPN based nanocomposites
Viscoelastic properties of all samples were determined with Dynamic Mechanical Analyzer Q800, TA instruments, USA. Samples of size 36 × 14 × 3 mm3, were analyzed at a fixed frequency of 1 Hz within a temperature range 30°C to 160°C with step size of 5°C/min using single cantilever mode.
Mechanical properties
Tensile strength of all samples was determined by using Instron 3366 UTM, USA at crosshead speed of 2 mm/min under load of 10 KN, using sample specifications according to ASTM D-638.
Results and discussion
Morphology
Figure 2a exhibits single phase morphology for pure VE and binary phase morphology for VE/silica nanocomposites. Bigger dispersed domains were observed in VES3 which indicates that nanoparticles, when added in higher amount, accumulated in some regions as the viscosity rose during curing reaction. For VES1 the sizes of aggregates are ranging from very small to big i.e. a wide size distribution is evidenced. However for VES2 mostly finer size of dispersed phase is observed with only a few small aggregates. Thus VES2 appears to have best dispersion of nano fillers in the resin among the three samples here.

(a) SEM of VE and VE/Silica nanocomposites. (b) SEM of 93VE IPN and 93VE IPN based nanocomposites.
Figure 2b shows morphology of the 93VE and 93VE based nanocomposites where the silica content was varied. Binary phase morphology is visible for all the samples except 93VE. Agglomeration of nanoparticles is more in 93VES1 and 93VES3 in comparison to that in 93VES2. Single phase morphology of 93VE and 93VES2 exhibits better miscibility of polymer components in the IPN and nanofiller/resin compatibility in the nanocomposite respectively.
Fourier transform infrared (FTIR) spectroscopy
The differences in the FTIR spectra (Figure 3a and b) of the vinyl ester resin and VES2 respectively indicate the types and extent of interactions between the matrix (functional groups at polymer chains) and the nanofiller. The broad band at 3417 cm–1, shown by circle, in Figure 3a, for cured VE resin, originates from hydroxyl group stretching vibrations and it was not visible in VES2.Simultaneously one small kink (shown by circle) appeared in the spectra of VES2 at 999 cm−1 (Figure 3b) which was due to stretching of Si-O-C.24,25 Thus it may be postulated that the -OH groups of VE were involved in chemical interaction with the silica filler during nanocomposite formation to form such Si-O-C linkage. The peak in the range of 2927–2866 cm−1 (shown by circle in Figure 3a) were due to CH3and CH2 stretching vibrations in VE resin and disappeared in the VES2 indicating the restricted mobility of these bonds due to newly formed covalent bonds between the VE and SiO2 (Figure 4). 25

FTIR spectra of VE and VSE2 nanocomposite (a) 4000-500 cm−1 (broad range) and (b) 2000-500 cm−1 (narrow range).

Interaction of VE with OMS nanoparticles.
However the nanocomposite formation did not cause much change in position of CH3 and CH2 bending vibrations corresponding to peaks at 1446 and 1361 cm−1 respectively. The peak at 1708 cm–1 was due to the stretching vibration of ester –O-C=O group present in vinyl ester resin which also appeared in VES2 (1713 cm–1) suggesting no major change in chemical structure of ester group.
Figure 5(i) shows comparison between FTIR spectra of VE neat resin, PU pre-polymer and cured VE/PU IPN (93VE). The characteristic peak of isocyanate groups at 2270 cm−1, in PU pre-polymer, disappeared when it was combined with vinyl ester resin in 93VE IPN and this may be attributed to the occurrence of chemical reactions between vinyl ester resin and PU (Figure 4). Gradual disappearance of the peak at 2270 cm−1 is accompanied by simultaneous reduction in intensity of the peak at 3448 cm−1 here. Reduction in peak depth at 3448 cm−1 compared to that in VE may be due to the involvement of –OH groups on VE in the reaction with –NCO of PU. 26

(i) FTIR spectra of (a) cured VE, (b) PU, (c) 93VE. (ii) FTIR spectra of 93VE IPN and 93VES2 nanocomposite.
In addition, the presence of the ester group corresponding to the peak position at 1720 cm−1, as was seen for both pure VE and PU pre-polymer in curve a and b respectively, remained unaltered after IPN formation (curve c). Also peak at 945 cm−1 for corresponding to C=C in VE, became insignificant in 93VE indicating that C=C in the VE reacted during IPN synthesis for crosslinking. Thus from FTIR study it is clear that two reactions had taken place simultaneously—i) addition reaction involving C=C and ii) rearrangement reaction involving NCO on PU and –OH on VE to form (-NHCOO-) urethane linkage. The peak at 1300 cm−1corresponds to urethane linkage (-NHCOO-) which is visible for the spectra of 93VE. 27
Figure 5(ii) exhibits FTIR spectra of silica nanofiller, 93VES2 and 93VE samples for comparison. Both 93VE and 93VES2 samples show almost similar spectra with unaltered peak depth and positions. However the peak at 559 cm−1 for nano-silica, which corresponds to Si-O-Si and O-Si-O bending, 28 contributed an effect in the spectra of 93VES2 (shown by circular region) though it was very less. The interaction between matrix resin and nanofiller through Si-O-C formation, as was postulated in VES2 according to Figure 4, may be considered here as the factor causing reduction in the peak intensity for Si-O-Si bending.
Specific gravity of nanocomposites
From the results of specific gravity (Tables 2 and 3) it is clear that i) with increase in silica filler content in the composites the difference between the experimental and theoretical specific gravity values increased and ii) with increase in filler content the specific gravity values of all the samples increased. Thus it may be concluded here that higher the percentage of filler more was the compactness in the composites which resulted greater specific gravity. Better compactness in the composites happens due to better interaction between resin and filler surfaces. 29 According to rule of additivity for a polymer blend of two components specific gravity may be expressed theoretically by the equation 2.
Variation of specific gravity of VE/OMS nanocomposites with OMS content (%weight).

Variation of specific gravity of 93VE IPN/OMS nanocomposites with OMS content (%weight).

However this equation is insufficient to explain the effect of filler content variation on specific gravity values of the composites based on such polymer blends as matrix in them. Further, to consider the effect of filler on the variation of specific gravity of composites equation 3 was proposed where filler, incorporated in the composites, was considered as a third component present in the medium and equation 3 was used to show the variation of the specific gravity of composites. 30 However, in the current study it was found that even after using this equation 3 the calculated values of specific gravity of actual composites was lower than that experimentally determined one.
So a new relation between the specific gravity and composition of composites based on polymer blends is necessary at this stage to have better understanding of the effect of filler content variation on polymer blends or IPNs. Based on the above data (Tables 2 and 3) the following relation may be written for the specific gravity (

Here n is positive integer whereas gn and wn are specific gravity and weight fraction of nth component in the composite respectively. When both x and Ṡ are positive quantities right hand side of equation 8 is higher than that of Equation 3 which explains the trends of experimental results observed in the present study. Quantitative value of Ṡ may be determined on the basis of the secondary valence forces between the interacting functional groups present on the filler and resin. 31 Greater the interaction between filler and resin more is the value of Ṡ. Experimentally observed specific gravity of specific composite was found higher than the expected value may be due to resin /filler interaction through Si-O-Si linkages.
Specific gravity of 93VE was higher than that of VE as was expected by rule of additivity. On the other hand, the lower specific gravity of 93VE based nanocomposites than that of VE based nanocomposites of corresponding composition, especially with higher filler content (≥2%), may be attributed to i) the reaction of the -NCO on PU chains to the reactive OH groups on VE resin during IPN formation, ii) lowering of the Si-O-Si interaction between resin /filler as was seen in VE/silica composites and iii) increase in the overall free volume. 25
Crosslink density
Figure 6a shows the effect of variation of filler content in nanocomposite on the crosslink density of VE matrix. Crosslink density of VE resin decreased with increase in silica content up to VES2 followed by increase for VES3. Lower crosslink density of nanocomposite than that of pure VE resin may be attributed to the interruption of curing reaction due to shielding of reactive functional groups (C=C) in VE pre-polymer by the nanoparticles. 25
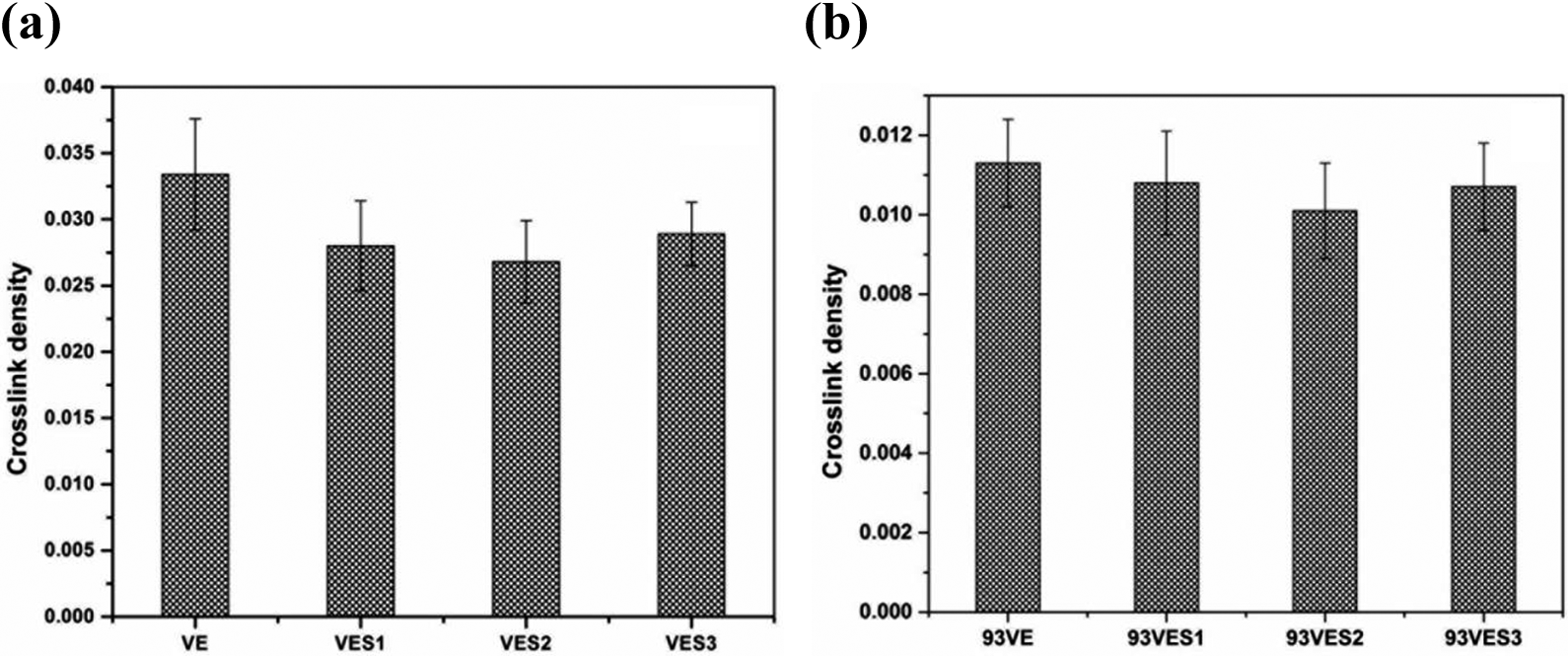
Variation of crosslink density of (a) VE/OMS nanocomposites and (b) 93VE based nanocomposites with OMS content.
With increase in silica content in the 93VE based composites crosslink density of the matrix reduced (Figure 6b) initially up to 2% silica content and then for 93VES3 again higher crosslink density was observed. At lower concentration of nanoparticles (≤2%) in the IPN, shielding of C=C &HO- functional groups on vinyl ester and NCO- on PU by the filler particles might have been dominated and hence hindrance for both the cross linking & grafting reaction as shown in Figure 7.With increase in silica content in 93VES3 and in VES3 agglomeration of the nanoparticles being dominated the functional groups on resin molecules were free to approach each other to react and hence the higher crosslinking density for 93VES3 and VES3.

Representation of plausible chemical reaction between VE and polyurethane during IPN formation.
Thermomechanical properties of VE/OMS and 93VE/OMS nanocomposite
Dynamic mechanical analysis (DMA) of VE/OMS nanocomposites was carried out to assess the thermal response of the samples under simultaneous application of dynamic load and heat in the range from 30°C to 160°C. All the samples showed very high storage moduli (Figure 8i.a) at 30°C. Neat VE showed highest storage modulus (∼19000 MPa) at room temperature. Incorporation of silica filler in VE caused reduction in initial storage modulus may be due to lower crosslink density after the intrusion of silica particles in the interstices of matrix. With increase in temperature there is gradual reduction in modulus except for VES2 which shows a plateau in the curve up to 60°C may be due to restricted segmental chain mobility in it. Such restricted chain mobility might have been caused by the predominant interaction between resin and filler (Figure 4) over the effect of crosslinking of the resin molecules. Figure 8b showed much less value of loss factor (tanδ) for the nanocomposites in comparison to that of neat resin which indicates that the resin/filler interaction had compensated the effect of lower crosslink density in the nanocomposites and restored the elasticity. Shifting of glass transition temperature, Tg, (i.e. temperature @ tanδmax) to higher values for the nanocomposites in the order VES < VES3 < VES2 was due to restricted chain mobility of the system due to the presence of nano fillers. VES2 sample may be considered as an optimum composition to have highest glass transition point (temperature at tanδmax) as well as elasticity among all the VE/OMS nanocomposites.

Variation of (i) (a) storage modulus, (b) tanδ of VE/OMS nanocomposites, (ii) (a) storage modulus, (b) tanδ of 93VE/OMS nanocomposites with temperature.
Viscoelastic properties of IPN based nanocomposites, namely storage modulus and tanδ, are plotted against the temperature and exhibited in Figure 8.ii(a) and (b) respectively. Storage modulus of all samples was high at room temperature though the nanocomposites showed lower value compared to that of the IPN. Also in the range of 40–80°C all the samples showed slow rate of fall in storage modulus. Rapid fall in storage modulus started beyond 80°C for nanocomposites and beyond 90°C for 93VE IPN (Figure 8.iia). Slower rate of fall in storage modulus with temperature for the IPN compared to that observed with the 93VE/OMS nanocomposites may be attributed to the higher degree of crosslinking in the former. According to Figure 8(ii)b loss tangent for the 93VE is lesser compared to that of 93VE/OMS nanocomposites suggesting that the former was comparatively more elastic than the nanocomposites. Also the Tg of IPN is greater than that of nanocomposites due to higher crosslink density of it.
Mechanical properties
It is evident from Figure 9 that the variation in %weight content of silica has influenced the mechanical properties of the nanocomposites. Mechanical properties of nanocomposites of vinyl ester with 1%, 2%, 3% and 5% silica by weight were evaluated. The composites exhibit higher mechanical properties, namely (a) tensile strength (b) tensile modulus and (c) toughness (Figure 9(i) a, b and c respectively) compared to that of neat cured vinyl ester resin matrix possibly due to the improved compatibility, through Si-O-C bonding at the interface of vinyl ester resin/silica. All the mechanical properties of VES2 are best among all the VE based nanocomposites studied here. Use of silica beyond 2% weight in the composites could not reinforce the matrix may be due to inefficient bonding between the filler and resin at the interface as filler particles were agglomerated and poor dispersion of these was seen in SEM pictures (Figure 2a). Highest toughness of VES2 (Figure 9.ic), among all the samples studied here, indicates the optimum balance between the rigidity through intra-molecular Si-O-C bonding and elasticity due to crosslinking of VE in it. 32 VES2 showed improvement in ultimate tensile strength by 83.5% and toughness by 42% compared to that of VE resin itself.

(i) Variation of (a) ultimate tensile strength, (b) tensile modulus, (c) toughness of VE/OMS nanocomposites with OMS content (%wt.), (ii) (a) ultimate tensile strength, (b) tensile modulus, (c) toughness of 93VE/OMS nanocomposites with OMS content (%wt.).
Tensile properties of IPN based nanocomposite (Figure 9.ii) e.g. UTS, tensile modulus and toughness were plotted against % OMS content in them. Tensile strength (Figure 9.iia) showed an increasing trend with increase in silica content up to 2 wt. % and then there was falling trend in property up to 93VES5. Modulus (Figure 9.iib) of all nanocomposites was inferior to 93VE IPN except 93VES2. Highest modulus for 93VES2 may be attributed to the increase in restricted segmental mobility of polymer matrix in presence of Si-O-C bonding with the silica nanofiller in the interstices in spite of its low degree of crosslinking. However with 93VES3 or 93VES5 such effect did not work probably due to predominated agglomeration of silica particles. Similarly toughness of nanocomposite (Figure 9.iic) showed a falling trend beyond 93VES2 after initial increase starting from 93VE. Highest toughness of the sample 93VES2 is in agreement with the lowest crosslink density of the system.
Also it is clear that 93VE IPN based nanocomposites possessed 2.5%, 16.47% and 20.98% higher tensile strength, modulus and toughness respectively than that of VE based nanocomposites of corresponding compositions.
Conclusion
Binary phase morphology of VE/OMS nanocomposites revealed that silica nanoparticles were not well dispersed in the vinyl ester resin matrices. Bigger aggregates were observed for VES3 but in VES2 finer size of dispersed domains indicated comparatively better mixing of filler with the resin. 93VES2 showed smaller dispersed domains than that seen in 93VES1 or 93VES3.
The broad band at 3417 cm–1, for cured VE resin, originated from hydroxyl group stretching vibrations, was not visible in VES2. Simultaneously one small kink appeared in the spectra of VES2 at 999 cm−1 which was due to stretching of Si-O-C. In VE based composite -OH groups on filler and resin were involved to form such Si-O-C linkage. Comparison of FTIR spectra of 93VE with that of VE showed remarkable reduction of the peak at 945 cm−1 (C=C) and 3448 cm−1 (OH) while simultaneous appearance of a new peak at 1300 cm−1(NHCOO-) in 93VE indicated possibility of reaction between VE and PU.
The use of silica beyond 2% weight in the composites could not reinforce the matrix may be due to inefficient bonding between the filler and resin at the interface as filler particles were agglomerated and poor dispersion of these was seen in SEM pictures.
Toughening of VE matrix due to IPN formation is well understood as the later showed lower storage modulus than neat VE. Also nanocomposites showed lower storage moduli at room temperature than 93VE and VE indicating more flexible nature of the composite than the matrix alone. The lower peak height in loss tangent vs. temperature curve for the IPN in comparison to that of 93VE/OMS nanocomposites and neat VE suggests that the IPN itself was comparatively more elastic than others. The order of variation of tanδmax was VE > 93VES2 > 93VE > VES2 nanocomposites and that of storage modulus in low temperature region was VE > 93VE > 93VES2 > VES2. Thus the sample 93VES2 may be considered as the optimum composition in respect of thermomechanical property.
From the overall analysis of properties of VE/OMS nanocomposites, 93VE IPN and 93VE/OMS nanocomposites, 93VES2 was found to be the optimum composition so far as the thermal stability, mechanical strength and elasticity of the matrix is concerned.
Footnotes
Acknowledgment
The authors are thankful to the Central Instrumentation Facility, BIT, Mesra for their help in the characterization of samples. The author also thanks IIT, BHU for allowing them to use SEM.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.