
Abstract
The synthesis and characterisation of two pyrrole monomers substituted at the N-position with a liquid crystal group are reported. Polymerisation of one of these compounds produced a soluble liquid crystalline semiconducting polymer. The side-chain of the polymer had a flexible spacer of nine methylene units, terminated by a mesogenic cyanobiphenyl group. The new compounds were chemically characterised and were found by hot-stage polarised optical microscopy to exhibit liquid crystal mesophases. The polymer was lightly doped p-type by exposure to iodine vapour, and its electrical conductivity was measured. The polymer synthesis and doping/dedoping processes were also briefly studied by cyclic voltammetry.
Introduction
There is widespread interest in the properties and potential applications of liquid crystalline conjugated polymers1–12, and pyrrole is one of the most promising building-blocks for the backbones of such polymers. However, the frequently-used 3-substituted pyrroles are nonsymmetrical monomers, effectively having a head (H) and a tail (T) 1 . Oxidative polymerisation of these molecules gives a somewhat random mixture of HH, HT, TT and TH connections between the repeating units of the polymer 1 . This causes steric crowding that will diminish the planarity and the order of the resulting polymer backbone, thus reducing its conductivity. In contrast, N-substituted pyrroles are effectively symmetrical; therefore the head and the tail are equivalent. The polymerisation of an N-substituted pyrrole (coupled regularly at the 2,5-positions) generates more regioregular coupling, thereby increasing the attainable conjugation of the polymer backbone1,4,7.
Better organisation might be produced by attaching a liquid crystal (LC) group to the backbone of the polymer1–7,10–12. In principle, this could improve the planarity and conjugation of the backbone, decrease the energy-gap and increase the mobility of the electronic charge-carriers 1 .
In this paper, we report the synthesis, polymerisation and properties of N-substituted pyrrole with a LC substituent, a strategy that has been little used1,4,5,9.
The mesomorphic properties of a liquid crystal are determined by the nature of the mesogenic group and the linking units 13 . Cyanobiphenyl, which has proven to be stable thermally and chemically, was chosen as the primary mesogen in this work. Our previous study used a six methylene-unit spacer to favour the nematic phase 14 ; in this work, a longer, nine methylene-unit spacer was chosen, in order to encourage the formation of a smectic phase 13 .
Experimental

The synthetic route for compounds 1, 2 and 3
Thermal optical microscopy was carried out using a NIKON Optiphot-2 polarising microscope in conjunction with a Linkam THMS 600 hot-stage and TMS 91 control unit. IR spectra were measured using a Perkin-Elmer Spectrum One ATR, and 1 HNMR spectra were recorded using JEOL Eclipse+ 400 FT-NMR (400 MHz for 1 H). The mass spectra were measured using a Varian 1200L Quadrupole MS. Differential scanning calorimetry (DSC) was performed on a Mettler DSC 25 instrument. A Keithley 224 current source and 617 programmable electrometer under computer control were used for 4-probe electrical conductivity measurements. X-ray diffractograms were obtained using a Bruker-AXS D-8 Advance powder X-ray diffractometer. The UV-visible spectra were measured using a Varian Cary 100 spectrophotometer.
4-Cyano-4'-hydroxybiphenyl (0.98g, 5 mmol) was added to a solution of 1,9-dibromononane (6.58 g, 23 mmol) and anhydrous potassium carbonate (3.17g, 23 mmol) in 2-butanone (60 mL); the mixture was refluxed for 21h. The reaction mixture was filtered and the solvent evaporated from the filtrate under reduced pressure. The residue was recrystallised from methylated spirit and purified by flash column chromatography (CHCl3 on silica). (Overall yield, based on 4-cyano-4'-hydroxybiphenyl: 1.12 g, 56%); mp 66°C. MS: m/z =399 and 401 (M+). FT-IR (KBr disk) vmax/cm−1: 2200 (CN), 1600 (C=C, Ar), 1245 (C-O-C).
1 H NMR (400 MHz, CDCl3): 8.00–8.10 (d, 2H, ArH); 7.50–7.60 (d, 4H, ArH); 7.49–7.36 (d, 2H, ArH); 3.93–4.01 (m, 2H, -CH2-O); 3.33–3.36 (t, 2H, -CH2-Br); 1.70–1.89 (m, 14H, -CH2-CH2-) ppm.
Synthesis of 1-bromo-9-(4-biphenyloxy-4'-carboxylic acid) nonane, Compound 2
To a mixture of water (5 mL), 95% sulphuric acid (2.7 mL) and glacial acetic acid (19.5 mL) was added 1-bromo-9-(4-cyano-4'-biphenyloxy)nonane (0.67g, 1.68 × 10−3 mol). The mixture was refluxed for 24 h, filtered, washed with distilled water and then cold industrial methylated spirit (IMS) before drying. The filtration residue was recrystallised 4 times from methylated spirit; yield: 0.22g, 30%. MS: m/z = 418 and 420 (M+). FT-IR (KBr disk) vmax/cm−1: 1601 (C=C, Ar); 1676 (C=O of COOH); 1245 (C-O-C). 1 H NMR (400 MHz, CDCl3) δ (ppm): 8.07–8.10 (d, 2H, ArH); 7.85–7.60 (d, 4H, ArH); 6.88–6.94 (d, 2H, ArH); 3.93–4.01 (m, 2H, CH2-O); 3.33–3.40 (t, 2H, CH2-Br); 1.71–1.83 (m, 14H, CH2-CH2).
Synthesis of 1-(N-pyrrole)-9-(4-cyano-4'-biphenyloxy) nonane, Compound 3
To a solution of 18-crown-6 (0.53 g, 2 mmol) in dry ether (100 mL) was added potassium t-butoxide (2.24 g, 20 mmol). A calcium chloride guard tube was fitted, the mixture was stirred at room temperature and pyrrole (1.4 g, 20 mmol) was added. Stirring was continued for 15 min. 1-bromo-9-(4-cyano-4'-biphenyloxy) nonane (Compound 1), (1.65 g, 4 mmol) in ether (20 mL) was added dropwise over a period of 10 min and the mixture was stirred for 50 h. Water (150 mL) was then added, and the mixture was extracted with ether (2 × 50 mL). The combined organic extracts were washed with saturated NaCl solution (50 mL), then water (50 mL) and dried over anhydrous MgSO4. The solvent was removed under reduced pressure, and the product was purified by column chromatography (CHCl3 on silica) and recrystallised from methylated spirit; yield: (0.74 g, 48 %). mp 115°C. MS: m/z = 386 (M+). FT-IR (KBr disk) vmax/cm−1: 2224 (CN), 1602 (C=C, Ar), 1251 (C-O-C). 1 H NMR (400 MHz, CDCl3) δ (ppm): 6.06–6.07 (t, 2H, pyrrole); 6.57–6.58 (t, 2H, pyrrole); 6.90–6.94 (d, 2H, ArH); 7.44–7.48 (d, 2H, ArH); 7.56–7.64 (m, 4H, ArH); 3.91–3.96 (t, 2H, CH2-O); 3.80–3.83 (t, 2H, CH2-N); 1.66–1.86 (m, 14H, CH2-CH2).
Synthesis of poly[N-nonyl-9-(4-cyano-4'-biphenyloxy)pyrrole]
The polymerisation of compound 3 was carried out by chemical oxidation under N2 atmosphere. The monomer (0.38 g, 1.0 mmol) was added to a filtered solution of anhydrous FeCl3 (0.32 g, 1.0 mmol) in CH2Cl2 (50 mL). The solution was heated at 30°C for 1h, and the oxidant residues were removed by extraction with sodium EDTA solution (2 × 50 mL, 0.1 M solution). The organic layer was washed with water (2 × 50 mL), dried (MgSO4), and the solvent was removed in a rotary evaporator under reduced pressure. The residual dark brown polymer was refluxed with methylated spirit and the solution was decanted to give a darker polymer residue and a soluble part (0.27 g). FT-IR (KBr disk) vmax/cm−1: 2224 (CN), 1604 (C=C, Ar), 1248 (C-O-C). 1 H NMR (400 MHz, CDCl3) δ (ppm): 6.07–6.08 (t, 2H, pyrrole); 6.57–6.58 (t, 2H, pyrrole); 6.91–6.93 (d, 2H, ArH); 7.45–7.48 (d, 2H, ArH); 7.56–7.64 (m, 4H, ArH); 3.91–3.95 (t, 2H, CH2-O); 3.81–3.84 (t, 2H, CH2-N); 1.66–1.83 (m, 14H, CH2-CH2).
Electrochemical polymerisation
The monomer (Compound 3) was electrochemically polymerised from a solution containing tetrabutylammonium tetrafluoroborate (TBAF; 0.14 g, 0.43 mmol) and the monomer (90 mg, 0.23 mmol) dissolved in 25 mL of dry propylene carbonate (N2 purged). The cell consisted of a stainless steel counter electrode, an indium-tin oxide (ITO) glass working electrode and a sealed saturated calomel reference electrode. The electrode potential was cycled 40 times from −0.3 V to +1.5 V vs. SCE, at a scan rate of 50 mVs- 1 .
Results and Discussion
Electrochemistry

A cyclic voltammogram showing the redox properties of the N-substituted pyrroles; scan rate 50 mV.s−1
In the first half-cycle, oxidative polymerisation of the monomer started at 1200 mV vs SCE; the subsequent reduction (dedoping) of the resulting orange polymer film was observed at about 600 mV. As would be expected, redoping of the polymer occurred at lower potential (ca. 750 mV) than that required for monomer oxidation. In the following oxidative half-cycle, the polymer film thickness (and hence the anodic current) increased. The colour of the film gradually changed to very dark brown as the cycling continued to form a coherent film of polymer.
The doping occurred at progressively higher potential in the successive oxidative stages, probably because the dedoped film resistance was becoming more significant after each cycle. In contrast, the cathodic dedoping potential remained quite constant because the oxidised films were reasonably conductive.
The electrochemical oxidation and reduction of the as-grown polymer occurred at higher potentials than that for polypyrrole (by ca. 350 mV). This is probably due to the steric effects of the un-aligned LC group, decreasing the effective conjugation length and hence increasing the ionisation potential of the polymer backbone.
Quantum Cache © software (Fujitsu Limited) was used to predict the optimised
local arrangement of the atoms in an isolated molecule of the monomer.

The N-substituted pyrrole monomer, Compound 3
On this simple basis, without consideration of intermolecular forces or the effects of polymerisation, the length of the monomer with LC group attached is 25.7 Å. After adding the van der Waals radii of the C≡N nitrogen and the pyrrole hydrogen (respectively 0.92 and 1.2 Å) to the length of the monomer, the effective length is 27.8 Å. This result is very close to the long spacing distance obtained by X-ray diffractometry (Section 3.5). The polymer was studied as well, and the results matched those of the isolated monomer.
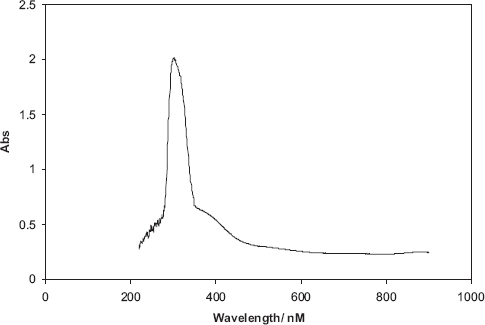
UV-visible spectrum of a dedoped film of the LC polymer on silica glass
However, after mild annealing of the sample at 135°C in vacuum for 2 h, the energy gap decreased to 2.6 eV; this is thought to be due to the self-organisation of the smectic liquid crystal phase, leading to a more planar backbone.
Films of the polymer were lightly doped by exposure to a mild oxidant, iodine vapour, for 24 h at ambient temperature. The electrical conductivity of this doped polymer was measured using the four-probe method to be 2.8 × 10−6 S m−1. This is less than that (1.3 × 10- 4 S m−1) of the analogous polymer with a shorter spacer 14 , which may be due to its lighter doping, or to the longer spacer increasing the steric hindrance.
The transition temperatures for all monomers and the polymer were studied by differential scanning calorimetry (DSC) and hot-stage polarising microscopy. For compounds 1 and 3, only a normal melting point was observed, These results are unlike those of a previous study of compounds analogous to 1 and 3 (with shorter spacers), which exhibited a nematic phase 14 . In the present work, Compound 2 (the acid) showed both a nematic and a smectic C phase, C→SC 144.8°C, SC→N 186.5°C, N→I 241.4°C, SC→C 147.0°C.
The nematic phase of compound 2 as it changed from isotropic to nematic was
homeotropic in appearance on a large proportion of the sample, and it flashed
when a mechanical pressure was applied to the slide. A polarised optical
micrograph texture is shown in

Polarised hot-stage micrograph of Compound 2 showing a bright appearance around the air bubbles in the homeotropic texture of the nematic phase at 229.1°C
Under the microscope, the phase changed again at 186.5°C to a schlieren texture.
It was identified to be a smectic C phase, since (a) the phase no longer flashed
and (b) the texture showed only four associated brushes, unlike the nematic
phase which shows centres with two brushes. The colour of the texture changed
with temperature from white to yellow-brown to blue on cooling. This is
consistent with the observations of Gray and Goodby
15
for the schlieren
texture of the smectic C phase with a colour change from yellow-grey to
yellow-brown and finally to blue, due to the tilt angle changes that occur as
the temperature decreases. The acid crystallised at 147.0°C. A micrographic
texture of the Sc phase is given in

Polarised hot-stage micrograph of compound 2 showing the schlieren texture of the smectic C phase at 179.1°C
The DSC trace of the polymer showed one peak, in addition to the glass
transition. By studying the polymer under hot-stage microscopy, a phase was
distinguished after holding the polymer at 136.3°C for 30 min. This phase was
further developed on holding the sample at 111.5°C for another 30 min. The
complex texture developed slowly, and it is believed to be due to a highly
organised liquid crystal mesophase; work is still in progress to characterise
it.

Polarised hot-stage micrograph of the polymer showing the texture exhibited after holding the sample at 136.3°C for 30 min

Polarised hot-stage micrograph of the polymer showing the texture that developed after holding the sample at 111.5°C for 30 min

X-ray diffractograms of the polymer as: [] a powder and [II] a pellet
XRD data for the LC polymer

The peak at 4.6
In this paper, the synthesis of new liquid crystal compounds is reported. The acid intermediate (product 2) showed two liquid crystal mesophases. A nematic and a smectic C mesophase were observed to be stable over a wide range of temperature. The pyrrole monomer was polymerised both chemically and electrochemically, and the redox properties of the resulting polymer were studied. The initially-formed polymer had a larger bandgap and lower conductivity than the previous one with a shorter spacer, but mild annealing of the polymer gave a lower energy gap due the LC self-organisation, and is likely to produce significantly increased conductivity. Modelling the monomer using Quantum Cache/Scigress software indicated molecular dimensions consistent with the repeat distances indicated by X-ray diffractometry. A well-organised texture was observed by polarised optical microscopy while holding the polymer at high temperature. In view of its relevance to the likely applications of LC conducting polymers17–19, the observed smectic mesophase is of more potential interest than the nematic one. Work is continuing to characterise the mesophase of the polymer fully, and to study the effects of an applied magnetic field and thermo-optic alignment on the organisation and the conductivity of the polymer.