
Abstract
Type 1 diabetes (T1D) is a chronic autoimmune disease characterized by the destruction of pancreatic beta cells (β-cell), leading to lifelong dependence on exogenous insulin. Despite advances in insulin delivery systems and glucose monitoring, achieving tight glycemic control remains a challenge for many patients. In recent years, stem cell–derived β-cell therapy has emerged as a promising strategy for restoring endogenous insulin production. This review explores the progress in differentiating human pluripotent stem cells, such as embryonic stem cells and induced pluripotent stem cells (iPSCs), into functional insulin-secreting β-like cells. We highlight recent breakthroughs in improving cell maturity, graft survival, and immune protection. Delivery approaches, encapsulation technologies, and transplantation strategies are discussed alongside an overview of ongoing clinical trials. The review also addresses current limitations, including immune rejection, cell dedifferentiation, and safety concerns. Finally, we examine future directions, such as genome editing, personalized cell therapy, and the integration of artificial intelligence in monitoring and optimizing treatment. Stem cell–derived β-cell therapy holds transformative potential as a curative approach for T1D, but further research is essential to overcome remaining barriers to its widespread clinical application.
Stem Cell–Derived β-Cell Replacement Therapy for Type 1 Diabetes: From Differentiation to Clinical Translation.
Keywords
Introduction
Type 1 diabetes (T1D) is a chronic autoimmune disorder characterized by immune-mediated destruction of pancreatic β-cells, resulting in absolute insulin deficiency and lifelong dependence on exogenous insulin 1 . Although insulin therapy has substantially improved glycemic management and patient survival, it fails to fully recapitulate physiological glucose homeostasis and is frequently associated with long-term microvascular and macrovascular complications, including nephropathy, neuropathy, and cardiovascular disease2,3.
Despite advancements in insulin formulations and delivery technologies, achieving long-term normoglycemia continues to be challenging for numerous patients, primarily due to the risks associated with hypoglycemia, glycemic variability, and chronic complications 4 Pancreatic islet transplantation demonstrates potential for restoring glycemic control in patients with T1D, although its broader clinical use remains constrained by donor scarcity, the need for immunosuppression, and the risk of graft attrition5,6.
Recent advancements in graft survival and immune protection have facilitated the clinical translation of stem cell–derived β-cell products, which are now progressing from preclinical studies into early-phase human trials distinct from conventional donor islet transplantation already practiced in several countries. Innovations such as new biomaterials, oxygenation-enhancing scaffolds, gene-edited hypoimmunogenic cell lines, and co-transplantation strategies are effectively addressing immune rejection and enhancing engraftment 7 . Recent work optimizing the transplantation microenvironment through improved vascularization, oxygenation, and biomaterial scaffolds has enhanced the engraftment and function of stem cell–derived β-cells, thereby expanding the range of clinically feasible transplantation strategies.
Over the past decade, stem cell–derived islet therapies have moved decisively from experimental proof-of-concept studies to early-phase clinical application. This transition is exemplified by several landmark first-in-human trials. ViaCyte has conducted multiple clinical studies evaluating pancreatic progenitor cells delivered within encapsulation devices, including PEC-Encap and PEC-Direct8,9, demonstrating safety, successful engraftment of stem cell–derived endocrine cells, and measurable C-peptide production in patients with T1D. More recently, Vertex Pharmaceuticals reported reproducible clinical benefit using fully differentiated stem cell–derived islets (VX-880), showing partial to complete insulin independence in multiple participants 10 . Together, these studies provide the first robust evidence that stem cell–derived islets can engraft, survive, and restore endogenous insulin production in humans, marking a significant step toward scalable cellular therapy for T1D.
Stem cell–derived β-cell therapy has emerged as a promising and potentially curative strategy for T1D by restoring endogenous insulin production through replacement of lost β-cell mass with functional insulin-secreting cells generated from human pluripotent stem cells, including hESCs and iPSCs11–14. Recent advances in directed differentiation, gene-editing technologies, and optimized culture systems have significantly improved β-cell yield, functional maturity, and glucose responsiveness. In parallel, innovations in immune protection and graft survival—such as encapsulation biomaterials, oxygenation-enhancing scaffolds, and hypoimmunogenic engineered cell lines—have further strengthened the translational potential and durability of stem cell–derived β-cell replacement therapies 15 .
Advances in developmental biology and the refinement of stepwise differentiation protocols have substantially improved the ability to generate pancreatic lineage cells from stem cells. These protocols recapitulate key stages of embryonic pancreatic development, enabling the production of β-like cells expressing hallmark markers such as insulin, pancreatic and duodenal homeobox 1 (PDX1), and V-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MAFA)12,16. Importantly, several clinical-grade stem cell–derived β-cell products have progressed into early-phase clinical trials, demonstrating initial safety and encouraging signals of metabolic efficacy 17 .
Despite this progress, significant challenges continue to limit widespread clinical adoption. These include incomplete functional maturation of β-like cells, immune-mediated rejection, potential tumorigenicity, and high manufacturing costs. In addition, optimization of transplantation routes, immune protection strategies, and long-term graft survival remains essential to achieving durable therapeutic benefit18–21.
This review synthesizes recent advances in stem cell differentiation efficiency, β-cell maturation, graft survival, and immune protection mechanisms. We critically evaluate contemporary delivery approaches, encapsulation and biomaterial technologies, and transplantation strategies, alongside evidence from ongoing clinical trials in T1D. In addition, the review examines current limitations, translational challenges, and emerging directions, including genome editing and artificial intelligence (AI)-enabled monitoring, that are shaping the future clinical adoption of stem cell–derived β-cell replacement therapies.
Figure 1a and b illustrates the chronological evolution of stem cell–derived β-cell therapy, highlighting key milestones from early pancreatic cell differentiation studies to current clinical trial breakthroughs.

Evolution of stem cell–derived beta-cell therapy.
Stem cell sources and differentiation into beta cells
Stem cell–derived β-cell replacement therapy aims to restore insulin secretion by generating mature, functional β-cells that recapitulate native pancreatic islet physiology, with human pluripotent stem cells serving as the principal translational platform 22 . Human embryonic stem cells (hESCs), first derived in 1998, represent the earliest renewable source of pancreatic progenitors and have demonstrated robust differentiation into insulin-producing cells through stepwise protocols that mimic embryonic pancreatic development23,24. Despite their genomic stability and differentiation efficiency, ethical and regulatory constraints limit widespread clinical use.
The clinical feasibility of cellular replacement was established by the Edmonton Protocol, pioneered by Dr A.M.J. Shapiro, which achieved insulin independence in patients with T1D following allogeneic islet transplantation under steroid-free immunosuppression, while simultaneously highlighting key limitations such as donor scarcity, variable graft durability, and lifelong immunosuppression25,26. Consequently, iPSCs have emerged as a complementary and potentially autologous alternative, offering scalable β-cell production with reduced immunogenic risk. Recent advances in gene editing and directed differentiation have further enhanced the functional maturation and translational potential of hESC- and iPSC-derived β-cells27–32.
Directed differentiation of hPSCs into pancreatic β-cells recapitulates embryonic development through sequential transitions from definitive endoderm to mature β-cells. Differentiation efficiency is dictated by precise temporal control of key signaling pathways, including Activin/Nodal, Wnt, retinoic acid, FGF, and Notch, and by coordinated transcription factor dynamics. Formation of a PDX1+/(NK6 homeobox 1) NKX6.1+ pancreatic progenitor population represents a critical checkpoint predicting β-cell yield and functional competence, while transient NEUROG3 induction governs endocrine commitment and lineage specification.
Despite progress, efficiency remains limited by intrinsic heterogeneity among hESC and iPSC lines and extrinsic constraints such as inadequate recapitulation of the pancreatic microenvironment, including morphogen gradients and oxygen availability. Three-dimensional culture systems, dynamic suspension platforms, and bioreactor-based approaches have demonstrated improved differentiation robustness and scalability by enhancing nutrient diffusion, mechanical signaling, and cellular uniformity.
Translational scalability remains a major challenge for stem cell–derived β-cell therapies. Although current protocols reliably generate insulin-expressing β-like cells, full functional maturation—defined by robust glucose-stimulated insulin secretion (GSIS), metabolic coupling, and long-term phenotypic stability—often requires extended in vitro culture or in vivo maturation. Large-scale Good Manufacturing Practice (GMP) manufacturing is further constrained by batch-to-batch variability, limited predictive potency assays, cryopreservation-associated functional loss, and the lack of standardized quality control metrics, underscoring critical barriers to clinical deployment.
In contrast, iPSCs, generated through somatic cell reprogramming using Yamanaka factors, offer a personalized and potentially autologous platform for β-cell production 33 . iPSC-derived pancreatic progenitors exhibit transcriptomic and functional profiles comparable to hESC-derived counterparts, while ongoing optimization of reprogramming fidelity and epigenetic memory aims to reduce inter-line variability—an essential requirement for scalable manufacturing and clinical translation34,35.
Collectively, these stem cell platforms reflect the transition from proof-of-concept differentiation systems toward GMP-compliant, clinically scalable β-cell sources. Nonetheless, persistent challenges related to uniform functional maturation, long-term stability, and cost-effective manufacturing remain. The establishment of standardized potency assays and differentiation benchmarks—such as C-peptide secretion thresholds and glucose stimulation indices—is critical for regulatory approval and inter-study comparability36,37.
Comparative overview of hESCs and hiPSCs
hESCs remain the benchmark for developmental fidelity and differentiation efficiency but are limited by ethical concerns and immune incompatibility. Human induced pluripotent stem cells (hiPSCs) offer an ethically acceptable and potentially autologous alternative; however, variability in reprogramming efficiency and epigenetic memory can affect reproducibility. Advances in genome editing and clonal selection strategies are increasingly mitigating these limitations and improving translational reliability38–40.
Advances in differentiation and maturation
Strategies for directing pluripotent stem cells toward a β-cell fate have evolved from two-dimensional monolayer cultures to advanced three-dimensional systems, including suspension bioreactors and pancreatic organoid models, which better recapitulate developmental and metabolic cues41,42. Contemporary protocols rely on the sequential modulation of developmental signaling pathways to guide cells through defined differentiation stages.
Key transcription factors—including PDX1, NKX6.1, NEUROG3, and MAFA—coordinate lineage commitment and β-cell identity, with MAFA expression closely associated with functional maturation and GSIS43,44. Optimization of culture conditions, particularly oxygen tension and metabolic support, has further enhanced β-like cell functionality, yielding near-physiological glucose responsiveness and improved post-transplant survival45,46.
Standardization and translational readiness
To support clinical translation, multiple academic and industrial groups are working toward standardized potency assays and real-time quality control frameworks for stem cell–derived β-cell products, with an emphasis on reproducible differentiation, batch release criteria, and long-term product stability47,48. Vertex Pharmaceuticals and ViaCyte are establishing standardized potency assays and real-time quality monitoring systems.
The development of functional β cells from stem cells represents a major milestone in the field of regenerative medicine for T1D. The most widely studied stem cell sources for this purpose include hESCs, iPSCs, and, to a lesser extent, mesenchymal stem/stromal cells (MSCs)49,50. Each of these sources has unique biological properties, advantages, and limitations in terms of scalability, immunogenicity, ethical considerations, and differentiation potential.
Human embryonic stem cells
hESCs were the first pluripotent cell source used to generate pancreatic progenitors due to their unlimited proliferative capacity and tri-lineage differentiation potential 11 . Developmentally guided protocols have successfully directed hESCs through definitive endoderm and pancreatic progenitor stages to insulin-expressing β-like cells 12 . However, concerns related to teratoma risk, ethical considerations, and immune incompatibility continue to limit their broad clinical application.
Induced pluripotent stem cells
Induced pluripotent stem cells (iPSCs), generated through somatic cell reprogramming, provide an ethically acceptable and potentially autologous alternative to hESCs, thereby reducing the risk of immune rejection2,19. iPSCs exhibit comparable pluripotency and differentiation capacity, and multiple studies have demonstrated efficient generation of insulin-producing β-like cells. Nonetheless, challenges related to inter-line heterogeneity and incomplete functional maturation remain key barriers to clinical translation 51 .
Mesenchymal stem/stromal cell encapsulation devices (MSCs)
Although MSCs lack the pluripotency of hESCs and iPSCs, they offer strong immunomodulatory and supportive functions. MSCs can enhance engraftment, reduce immune responses, and serve as co-transplanted helper cells52,53. While they are not a direct source of insulin-secreting cells, their role in islet niche regeneration and immune tolerance is increasingly recognized.
Adult stem/progenitor cells
Adult stem/progenitor cells derived from pancreatic or extra-pancreatic tissues have been explored for their potential to support β-cell regeneration. Although their plasticity is limited compared with pluripotent stem cells, these populations may contribute indirectly through paracrine signaling or, under defined injury or inflammatory conditions, transient expression of endocrine lineage markers52,54–56. However, their capacity to generate functionally mature β-cells remains insufficient for standalone therapeutic application.
MSCs, a subset of adult stem cells, exert supportive effects by modulating immune responses, reducing fibrosis, and promoting vascularization at the graft site53,57. Consequently, MSC co-transplantation has been proposed as an adjunctive strategy to enhance engraftment, survival, and functional stability of stem cell–derived β-cell therapies.
Sequential differentiation of hPSCs into pancreatic β-cells is orchestrated by stage-specific signaling pathways and transcriptional programs (Table 1). Each transition involves characteristic molecular markers and functional benchmarks used to assess lineage fidelity and maturation.
Key signaling pathways, stage-specific markers, and functional notes in directed differentiation of hPSCs into pancreatic β cells.

Mechanistic and practical considerations
Precise timing and dosage of growth factor signaling critically determine lineage specification and β-cell yield, with NKX6.1 induction at the pancreatic progenitor stage serving as a key predictor of functional β-cell output. Three-dimensional culture systems including bioreactors, organoids, and extracellular matrix–based platforms enhance maturation by better recapitulating the native microenvironment. Robust quality control, supported by standardized potency assays and quantitative functional benchmarks, is essential for inter-study comparability and clinical translation. Recent advances have enabled the generation of glucose-responsive β-like cells from hESCs and iPSCs, culminating in the first reported human transplantation of iPSC-derived islets, underscoring the translational potential of pluripotent stem cell–based approaches beyond donor islet replacement8,58.
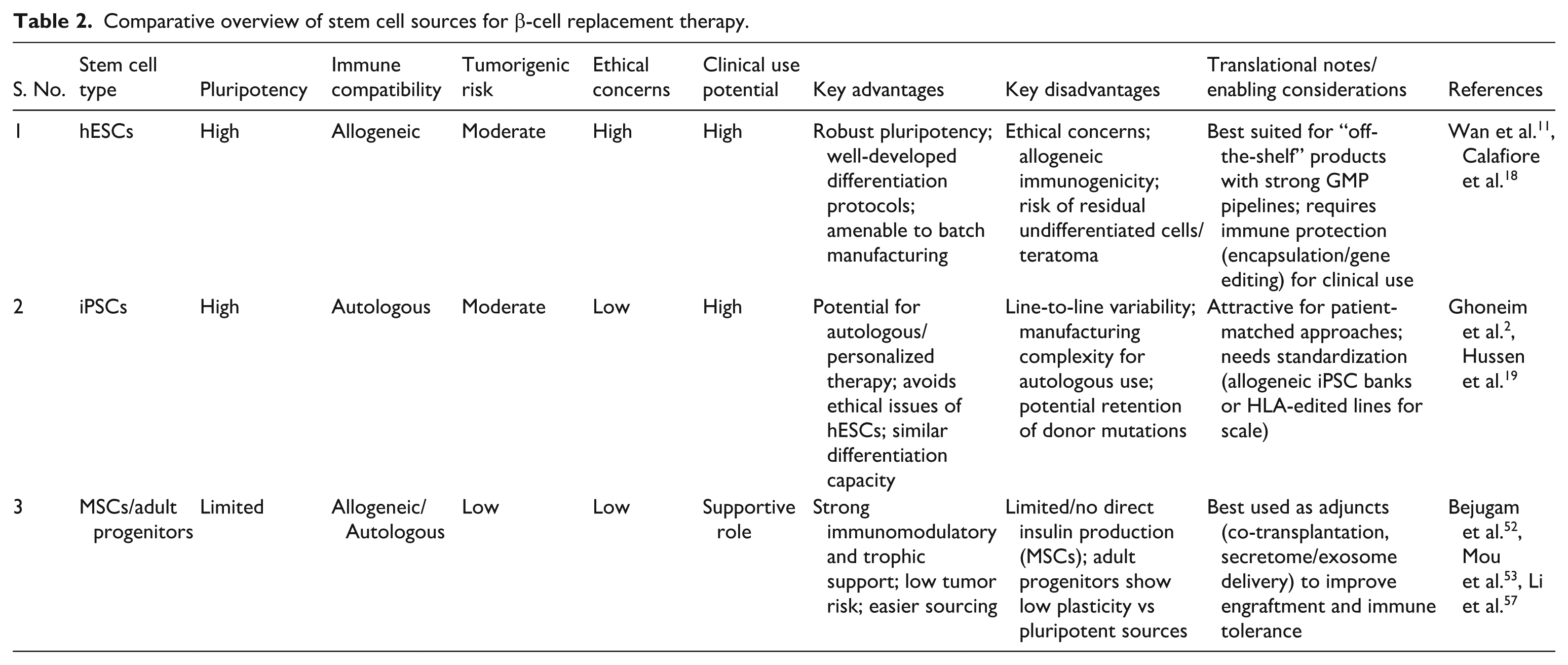
Table 2 contrasts the major stem cell sources (hESCs, iPSCs, and adult progenitors such as MSCs) in terms of translational strengths and limitations. hESCs remain the most mature platform but face ethical and immune barriers; iPSCs offer personalization yet require standardization and safety validation; MSCs and adult progenitors have supportive roles rather than direct insulin replacement. Together, these sources highlight the trade-off between scalability, immunogenicity, and therapeutic function.
Comparative overview of stem cell sources for β-cell replacement therapy.
Functional maturation and
in vitro
assessment
Achieving full maturation of stem cell–derived β-like cells is essential for their clinical success. Although differentiation protocols can generate insulin-producing cells, many lack the full functionality of native pancreatic β cells. This section highlights the key markers and methods used to evaluate their maturity and performance.
Maturity markers of β-cell
Mature β cells are characterized by the expression of specific transcription factors and proteins, including:
The co-expression of these markers suggests a more functionally competent β-like cell population12,18.
Functional testing methods
Functional competence of stem cell–derived β-like cells is most commonly evaluated using GSIS assays. In this approach, cells are sequentially exposed to low and high glucose concentrations, and insulin or C-peptide release is quantified. A reproducible, glucose dose–dependent increase in secretion is considered a hallmark of functional β-cell maturation 62 .
In addition, calcium flux assays are employed to assess stimulus–secretion coupling. Glucose-induced intracellular calcium influx reflects appropriate membrane depolarization and activation of voltage-gated calcium channels, providing complementary evidence of glucose responsiveness and secretory readiness.
Comparison with native β-cell
Despite substantial progress, stem cell–derived β-like cells often do not fully match the functional profile of native pancreatic β-cells. Compared with primary human islets, they may exhibit reduced insulin secretory capacity, delayed glucose responsiveness, and residual expression of immature or polyhormonal markers2,16. These differences highlight incomplete metabolic and electrophysiological maturation.
Functional benchmarks such as GSIS and calcium flux imaging remain the primary assays for direct comparison with native β-cells. Importantly, recent studies have shown that human iPSC-derived β-cells cultured or encapsulated within vascularized and biomimetic matrices display improved insulin secretion dynamics and glucose responsiveness compared with unstructured aggregates, underscoring the critical role of the microenvironment in achieving near-physiological function 63 .
Delivery methods and transplantation strategies
The success of stem cell–derived β-cell therapy depends not only on generating functional cells but also on delivering them effectively into the patient. Selecting the appropriate transplantation route and providing a protective environment are crucial to maximize graft survival, functionality, and safety.
Transplantation sites
Several anatomical sites have been explored for cell transplantation.
The choice of transplantation site is a key determinant of graft survival, oxygenation, and long-term function of stem cell–derived β-cells. The intraportal hepatic route, established as the clinical standard following the Edmonton protocol, offers rapid vascularization and relatively high oxygen tension (≈80–100 mm Hg) with direct access to portal glucose flux 64 . However, this site is associated with significant limitations, including instant blood-mediated inflammatory reaction (IBMIR), early inflammatory injury, microvascular embolization, limited graft retrievability, and progressive loss of graft mass over time. These challenges have driven increasing interest in alternative extrahepatic transplantation sites that may better support engraftment, immune protection, and long-term functional stability.
Subcutaneous implantation is attractive because of its procedural simplicity, safety, and graft retrievability, and has therefore been adopted in several stem cell–based clinical programs. However, its major limitation is poor oxygen availability, with reported oxygen tensions substantially lower than intraportal or omental sites. Inadequate vascularization creates a hypoxic microenvironment that compromises early engraftment, β-cell survival, and insulin secretory capacity, ultimately reducing long-term graft viability.
To address these constraints, recent studies have focused on pro-angiogenic and oxygen-modulating strategies. Biomaterials incorporating controlled-release vascular endothelial growth factor (VEGF), oxygen-releasing scaffolds, and pre-vascularized matrices have been shown to enhance local perfusion, mitigate ischemic stress, and improve functional outcomes of subcutaneous grafts65–72. These approaches highlight the importance of microenvironmental engineering in overcoming site-specific limitations.
Notably, recent clinical programs led by ViaCyte and Shapiro’s group have renewed interest in the subcutaneous space by demonstrating the feasibility of implanting encapsulated, stem cell–derived islet cells. While these trials confirm procedural safety and retrievability, they also underscore persistent challenges related to hypoxia and delayed vascular integration, which remain key barriers to durable glycemic efficacy73,74.
Emerging strategies, including oxygen-permeable biomaterials, VEGF-releasing scaffolds, and co-transplantation with MSCs, are being actively investigated to enhance local oxygenation, promote vascular integration, and improve graft survival, underscoring oxygen supply as a critical determinant of long-term clinical success 35 . In parallel, alternative anatomical transplantation sites are being explored to optimize islet viability and retrievability 75 . Although the subcutaneous space offers procedural simplicity and accessibility, it is limited by poor vascularization and restricted oxygen diffusion, which negatively affect graft longevity. To mitigate these constraints, pre-vascularized and oxygen-enhancing scaffolds, as well as encapsulation devices, have been developed to improve local perfusion while providing immune protection. In addition, the omental pouch—owing to its rich vascular network and favorable surgical access—has gained renewed interest as a transplantation site capable of supporting rapid revascularization and sustained glycemic control76–78.
The omentum and liver surface have emerged as promising alternative transplantation sites that combine improved oxygenation with favorable vascular and immunological profiles. Notably, Endo Kumata et al. 66 described a novel liver-surface transplantation strategy in which islets are implanted onto the hepatic capsule using a biodegradable gelatin hydrogel scaffold. This approach preserves metabolic integration while enhancing local oxygenation and minimizing exposure to the IBMIR, thereby supporting rapid revascularization and sustained insulin secretion without major complications 66 . In parallel, other anatomical niches—including the renal subcapsular space, intramuscular sites, and bone marrow—are being explored for their potential to support islet survival and function, though clinical evidence remains limited75,79.
Overall, transplantation site selection is a critical determinant of therapeutic efficacy. While subcutaneous sites offer procedural simplicity and retrievability, their intrinsic hypoxia necessitates biomaterial-based augmentation. In contrast, scaffold-assisted and liver-surface strategies underscore the importance of integrating site-specific physiology with engineered microenvironments to optimize β-cell engraftment, function, and long-term clinical outcomes.
Encapsulation and delivery technologies
Encapsulation technologies are critical for the clinical translation of stem cell–derived β-cell therapies, providing immune protection while maintaining graft viability and function. These systems create a semi-permeable microenvironment that permits nutrient, oxygen, and insulin exchange while limiting host immune recognition. Encapsulation strategies broadly include macroencapsulation and microencapsulation, each with distinct advantages and constraints67,80.
Macroencapsulation systems
Macroencapsulation platforms, such as planar or pouch-based devices, house large cell populations within retrievable implants, enabling physical immunoisolation and post-transplant monitoring. Devices exemplified by ViaCyte’s Encaptra system facilitate surgical retrieval but are limited by oxygen and nutrient diffusion barriers. Experimental studies indicate effective oxygen diffusion is restricted to approximately 100–200 μm, beyond which hypoxia compromises β-cell survival and insulin secretory kinetics 81 . To mitigate these limitations, recent designs incorporate oxygen-permeable membranes, reduced device thickness, prevascularization strategies, and angiogenic surface modifications to enhance local perfusion.
Microencapsulation strategies
Microencapsulation strategies embed individual islets or small β-cell clusters within hydrogel microspheres, typically alginate-based, offering improved mass transfer due to a high surface-area-to-volume ratio. This configuration supports rapid glucose sensing and insulin secretion but is susceptible to pericapsular fibrotic overgrowth driven by foreign body responses, ultimately impairing long-term graft function. Advances in biomaterial chemistry—including ultra-purified and chemically modified alginates, zwitterionic polymers, and optimized cross-linking—have demonstrated reduced macrophage adhesion and fibrosis in preclinical models. In addition, co-encapsulation with immunomodulatory agents or mesenchymal stromal cells has shown promise in extending graft survival 66 .
Oxygenation and pro-angiogenic scaffolds
Inadequate oxygenation remains a critical limitation for encapsulated β-cell grafts, particularly at avascular implantation sites. To mitigate hypoxia, oxygen-generating biomaterials and pro-angiogenic scaffolds have been developed to enhance local oxygen delivery and accelerate revascularization. Endo Kumata et al. 66 demonstrated that liver-surface islet transplantation using a biodegradable gelatin hydrogel scaffold improved oxygenation, vascular integration, and sustained insulin secretion. Emerging hybrid biomaterials that combine extracellular matrix components, angiogenic cues, and optimized mechanical properties further promote β-cell survival and maturation. Rational scaffold design guided by diffusion kinetics and β-cell biology is essential for achieving durable and clinically scalable β-cell replacement therapies.
Comparative overview
Table 3 presents a comparison of current encapsulation and delivery platforms, highlighting their strengths, limitations, and enabling technologies. Macrophages offer clinical retrievability and controlled dosing, microcapsules provide superior diffusion dynamics and modular scalability, and oxygenation scaffolds integrate metabolic support into the graft environment. Recent innovations in encapsulation materials and scaffold engineering are bridging the gap between biological function and immune protection, with continued refinement of oxygen diffusion properties and vascular integration crucial for clinically durable and immune-protected therapies.
Comparative overview of transplantation sites and cell-delivery devices.

Table 3 summarizes current and emerging transplantation sites for stem cell–derived β-cell therapy, emphasizing their relative strengths and challenges. While the intraportal route remains the clinical benchmark, alternative sites such as the omentum, subcutaneous space, and liver-surface scaffolds offer improved accessibility and reduced immune activation. Notably, the recent study by Endo Kumata et al. 66 demonstrated successful islet engraftment on the hepatic surface using a gelatin hydrogel scaffold, representing a promising extrahepatic strategy. Advances in biomaterials, encapsulation devices, and pre-vascularized matrices continue to enhance graft viability, paving the way for safer and more efficient transplantation approaches.
Scaffold-based and hydrogel platforms
Scaffold-based and hydrogel platforms support three-dimensional β-cell organization, enhance vascular integration, and mimic key extracellular matrix cues, thereby improving graft viability and functional stability 57 . Materials such as alginate, collagen, and polyethylene glycol (PEG)–based hydrogels offer tunable mechanical and diffusional properties and are well suited for extrahepatic sites, including the omentum and subcutaneous space. Beyond intraportal delivery, Endo Kumata et al. 66 demonstrated effective liver-surface islet transplantation using a biodegradable gelatin hydrogel scaffold, achieving improved revascularization and sustained graft function.
Figure 2 depicts the different transplantation sites (liver, omentum, subcutaneous) alongside encapsulation technologies (macro- and microcapsules) and hydrogel-based delivery platforms.

Delivery routes and encapsulation devices.
Oxygenation and pro-angiogenic strategies
Recent advancements in subcutaneous grafts have focused on overcoming the oxygenation barrier using oxygen-releasing biomaterials, microvascularized scaffolds, and pro-angiogenic delivery systems. Hydrogels loaded with calcium peroxide or hemoglobin-based oxygen carriers can sustain localized oxygen tension, improving β-cell survival during early engraftment. VEGF-functionalized matrices and pre-vascularized scaffolds enhance microvessel formation and faster integration with host vasculature66,80,82–84.
Immunological challenges and evasion strategies
Immune rejection remains a major barrier to stem cell–derived β-cell therapy, affecting both allogeneic and autologous grafts through alloimmunity, recurrent autoimmunity, and foreign body responses. Recent advances demonstrate that gene-editing approaches, including PD-L1 overexpression and HLA class I deletion, combined with localized immune-modulatory biomaterials such as SA-FasL microgels, can promote long-term graft survival without systemic immunosuppression 15 . Integrating intrinsic hypoimmunogenicity with extrinsic immune cloaking represents a promising strategy for durable immune tolerance.
Allogeneic versus autologous transplantation
Autologous transplantation, where cells are derived from the patient’s own tissue (e.g., using iPSCs), minimizes immune rejection but is expensive, time-consuming, and may still carry autoimmune risk in T1D patients11,18.
Host immune response and rejection
Transplanted cells face both innate and adaptive immune responses:
Innate immunity may trigger inflammation immediately post-transplant.
Adaptive immunity, particularly T-cell-mediated cytotoxicity, poses long-term risks of graft loss, especially in HLA-mismatched allogeneic therapies 20 .
Autoimmune responses against β-cell antigens may also resurface, particularly in T1D patients.
Strategies for immune evasion
Immunosuppression versus Immune Cloaking
Systemic immunosuppressants (e.g., tacrolimus, sirolimus) are traditionally used but come with serious side effects like infections and organ toxicity 86 .
Immune cloaking is a promising alternative. This includes: ○ Gene-editing technologies, such as HLA knockout or PD-L1 overexpression, to make transplanted cells “invisible” to the immune system without dampening overall immunity63,87. ○ Co-transplantation with immunomodulatory cells, like MSCs, which secrete anti-inflammatory cytokines and promote immune tolerance52,53.
These strategies may reduce or eliminate the need for systemic immunosuppressants, improving safety and patient outcomes.
Immune evasion strategies in islet transplantation
Immune rejection remains a critical barrier to durable β-cell replacement in T1D, driven by alloimmune responses and recurrent autoimmunity. Conventional systemic immunosuppression is associated with substantial risks, including infection, nephrotoxicity, and malignancy, prompting the development of graft-intrinsic immune evasion strategies 88 .
Recent advances in genome editing have enabled the generation of hypoimmunogenic stem cell–derived β-cells. Targeted disruption of human leukocyte antigen (HLA) class I and II molecules reduces T cell–mediated allorecognition; however, complete HLA ablation may increase susceptibility to natural killer (NK) cell–mediated cytotoxicity. To counter this, combinatorial strategies incorporating PD-L1 overexpression and CD47 expression have been developed. PD-L1 suppresses T-cell activation via the PD-1 pathway, while CD47 delivers a “don’t-eat-me” signal to innate immune cells. Preclinical studies using multiplex Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) editing demonstrate prolonged graft survival without systemic immunosuppression, supporting the feasibility of immune-invisible grafts80,89,90.
Complementary immune cloaking technologies, including encapsulation within biocompatible hydrogels and nanoscale biomimetic coatings, physically restrict immune cell infiltration while preserving nutrient and insulin diffusion 66 . In addition, co-transplantation with MSCs enhances graft survival by reducing inflammation, promoting vascularization, and inducing regulatory T-cell responses. Early clinical studies suggest safety and feasibility, though long-term efficacy remains under evaluation74,91,92.
Immune protection and engineering
Immune rejection remains a major barrier to durable engraftment of transplanted β-cells, while conventional systemic immunosuppression is associated with significant risks, including infection, nephrotoxicity, and malignancy 93 . Consequently, contemporary strategies emphasize graft-intrinsic and localized immune modulation to reduce or eliminate the need for lifelong systemic therapy.
Gene editing for hypoimmunogenic cells
Gene-editing technologies, particularly CRISPR–Cas systems, enable targeted modification of immune-recognition pathways to generate hypoimmunogenic stem cell–derived β-cells. Disruption of HLA class I and II molecules reduces CD8+ and CD4+ T-cell–mediated allorecognition, significantly prolonging graft survival in preclinical models 66 . However, complete HLA ablation increases susceptibility to NK cell–mediated cytotoxicity. To address this, HLA editing is often combined with expression of non-classical HLA molecules (e.g., HLA-E) or immune-inhibitory ligands such as PD-L1 and CD47. PD-L1 suppresses T-cell activation through checkpoint signaling, while CD47 inhibits innate immune clearance, collectively enhancing immune tolerance beyond single-modality approaches.
Biomaterial-based immune cloaking
Biomaterial-based immune cloaking complements genetic strategies by creating localized immune-privileged environments. Encapsulation membranes, nano-porous barriers, and biomimetic coatings restrict immune cell infiltration while preserving oxygen, nutrient, and insulin diffusion. Recent advances incorporating tolerogenic ligands and anti-fibrotic materials have demonstrated reduced inflammatory responses and improved graft durability in preclinical studies52,53.
Cellular co-therapies
MSCs provide potent immunomodulatory support by secreting anti-inflammatory cytokines, inducing regulatory T cells, and promoting angiogenesis. Co-transplantation of MSCs or MSC-derived extracellular vesicles has enhanced graft survival and function in preclinical and early clinical studies8,80, particularly when integrated with gene-edited or encapsulated grafts.
Translational and regulatory considerations
Emerging consensus supports multimodal immune engineering, combining gene biomaterial cloaking, and cellular co-therapies to achieve durable tolerance. However, clinical translation requires rigorous evaluation of genomic integrity, off-target effects, oncogenic risk, and long-term phenotypic stability. Harmonized regulatory frameworks, scalable manufacturing, and extended post-transplant surveillance remain essential before routine clinical adoption5,8,20,25,66,80.
Clinical trials and translational milestones
Over the past decade, stem cell–derived β-cell replacement therapy has advanced from preclinical proof of concept to early clinical evaluation in patients with T1D. Recent trials by ViaCyte and Vertex, along with emerging Chinese studies using CiPSC-derived islet-like cells, have demonstrated restoration of insulin secretion, partial to complete insulin independence, and favorable safety profiles. Encapsulated stem cell–derived islet grafts have achieved sustained glycemic control for up to 1 year, marking a critical step toward clinically viable β-cell replacement therapies.
The Edmonton Protocol, designed by Dr A.M.J. Shapiro, transformed islet transplantation by enabling insulin independence for patients with T1D without the use of steroids. This protocol, corroborated by multicenter investigations, establishes the benchmark for beta-cell replacement techniques. Recent breakthroughs, such as clinical-grade CiPSC-derived islet-like cells and novel transplantation procedures, indicate significant translational success. A comparative analysis of these trials, ranging from the Edmonton Protocol to contemporary stem cell therapies, is illustrated in Table 4.
Major clinical trials in stem cell–derived beta-cell therapy for type 1 diabetes.

Table 4 summarizes key clinical trials investigating stem cell–derived beta-cell therapies for T1D. It outlines the study design, registration details, indication, trial phase and current status, major objectives, endpoints, limitations, and references. This comparative overview highlights the translational progress, safety considerations, scalability issues, and regulatory status (e.g., FDA or Country Specific regulatory authority involvement), offering insights into the evolving clinical landscape of cellular therapies.
Key clinical outcomes
i. Vertex Pharmaceuticals (VX-880 and VX-264 Trials).
Vertex’s VX-880 program represents the first clinical application of stem cell–derived pancreatic endoderm cells delivered via the intraportal route under standard immunosuppression. Early Phase I/II results demonstrated robust restoration of endogenous insulin secretion, with marked increases in C-peptide levels, >50% reduction in exogenous insulin requirements in several participants, and insulin independence achieved in a subset within 12 months. Graft function has persisted beyond 24 months in early recipients, with no evidence of teratoma formation or off-target differentiation. Adverse events were primarily related to immunosuppressive therapy and included transient hepatic enzyme elevations. The follow-on VX-264 program incorporates a macroencapsulation device designed to eliminate the need for systemic immunosuppression. Preclinical data indicate preserved insulin secretion with minimal immune infiltration, although long-term clinical engraftment outcomes remain under evaluation. Together, these programs illustrate a transition from open transplantation toward encapsulated, retrievable cell therapies.
ii. ViaCyte’s PEC-Encap device achieved pancreatic progenitor engraftment but was limited by fibrotic overgrowth and oxygen diffusion constraints. In contrast, the PEC-Direct system, which permits vascular ingrowth under immunosuppression, demonstrated detectable human C-peptide in over 60% of recipients within 6–12 months. These findings have informed the development of next-generation devices incorporating oxygen-permeable membranes and angiogenic coatings.
iii. In a Chinese clinical trial by Wang et al. 35 , one participant with longstanding T1D received autologous iPSC-derived β-like clusters implanted intramuscularly without chronic immunosuppression. The results showed C-peptide restoration in patient and an average HbA1c reduction of 1.2% over 9 months. No tumorigenic lesions were found during imaging or biopsy, indicating the safety and partial efficacy of personalized iPSC-based grafts, despite the absence of insulin independence.
iv. Regulatory and Safety Considerations
First-in-human studies must adhere to strict Investigational New Drug (IND) frameworks set by the Food and Drug Administration (FDA), National Medical Products Administration (NMPA), and European Medicines Agency (EMA), focusing on product consistency, potency assays, and long-term follow-up. Key safety endpoints include tumorigenicity, off-target differentiation, and immune sensitization, requiring post-transplant surveillance for over 5 years. In addition, harmonizing GMP standards and transparent reporting of adverse events are essential for regulatory convergence and market authorization. Recent, clinical trials show that stem cell–derived β-cell replacement therapies are achieving functional proof-of-concept in humans, with notable improvements in insulin production
94
. Progress in encapsulation and immune engineering indicates advances toward a curative therapy for T1D. Nevertheless, challenges such as long-term graft stability, immune protection without systemic immunosuppression, and scalable manufacturing need ongoing collaboration among academia, industry, and regulatory bodies.
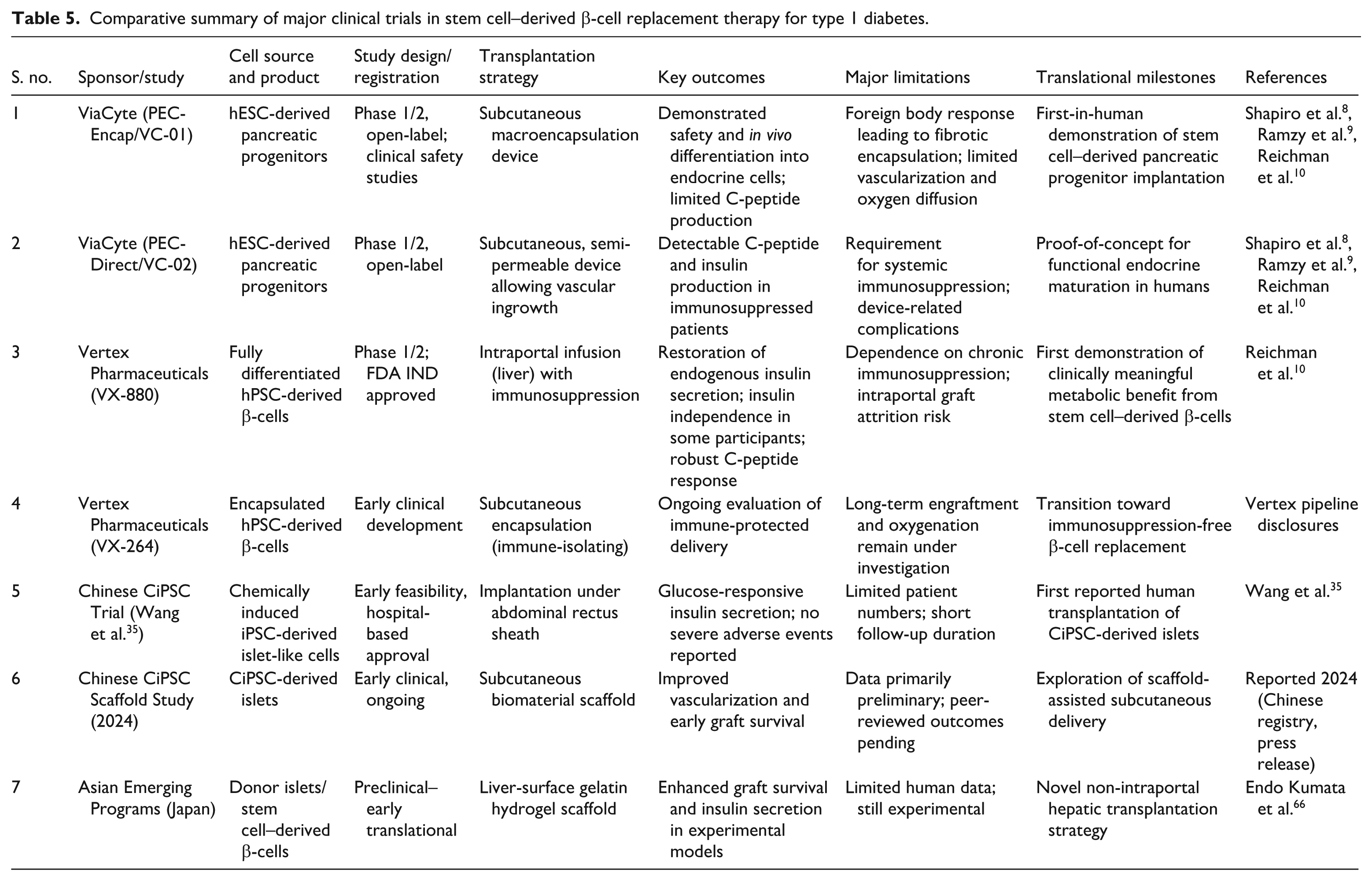
Collectively, these clinical programs (Table 5) illustrate a clear progression from safety-focused implantation of stem cell–derived progenitors toward functionally mature β-cell replacement with clinically meaningful metabolic outcomes. ViaCyte trials established feasibility and safety but highlighted challenges related to fibrotic encapsulation and oxygen diffusion. Vertex’s VX-880 trial represents a pivotal milestone, demonstrating insulin independence using fully differentiated β-cells, albeit with reliance on systemic immunosuppression. Emerging CiPSC-based studies in Asia introduce alternative reprogramming strategies and innovative transplantation sites, underscoring global diversification of translational approaches. Despite encouraging outcomes, key limitations—including immune protection without immunosuppression, long-term durability, and scalable manufacturing—remain central to future clinical advancement.
Comparative summary of major clinical trials in stem cell–derived β-cell replacement therapy for type 1 diabetes.
Limitations and translational implications
Across clinical trials, immune rejection remains a major limitation, often necessitating systemic immunosuppression with associated safety risks. Although short-term insulin independence and improved glycemic control have been achieved, long-term graft durability remains inconsistent, with declining function reported in some studies. Subcutaneous and omental transplantation sites are further constrained by limited vascularization and low oxygen tension, adversely affecting engraftment. Variability in differentiation protocols also raises concerns regarding mixed cell populations and residual tumorigenic risk.
Future efforts focus on biomaterial-assisted transplantation to enhance oxygenation, gene-editing strategies such as HLA modification to reduce immunogenicity, and improved encapsulation platforms. However, scalable manufacturing, regulatory harmonization, cost, and ethical considerations remain key challenges for widespread clinical adoption.
Limitations and current challenges
Stem cell–derived β-cell therapy shows strong translational promise but faces substantial challenges related to scalability, cost, and regulatory implementation. Long-term clinical success will depend not only on biological efficacy but also on the development of economically viable, ethically acceptable, and reproducible manufacturing pipelines.
Manufacturing and GMP production constraints
Clinical deployment requires GMP-compliant differentiation systems capable of consistently producing functional insulin-secreting β-cells at scale 48 . Current protocols remain labor-intensive, often exceeding 30 days per batch, with variable yields and production costs estimated at USD 8000–15,000 per million cells. While automation through closed-system bioreactors and xeno-free media may improve reproducibility, meeting population-level demand remains a major bottleneck.
Cryopreservation and supply chain logistics
Cryopreservation of β-like cells is limited by post-thaw loss of viability and function due to membrane damage and oxidative stress. Advances in cryoprotectant formulations, controlled-rate freezing, and real-time cold-chain monitoring are essential to maintain product integrity during distribution 95 . Regional biobanking and decentralized manufacturing may further improve accessibility and reduce logistical costs.
Economic modeling and cost-effectiveness
Estimated treatment costs range from USD 200,000 to 400,000 per patient, comparable to pancreas or islet transplantation96,97. However, long-term insulin independence could offset costs by reducing complications such as hypoglycemia, nephropathy, and retinopathy, potentially yielding acceptable cost-effectiveness ratios over time 98 . Ethical considerations—particularly regarding hESC use—have accelerated the shift toward iPSC-based platforms. Clear reimbursement models, value-based payment frameworks, and transparent regulatory oversight will be essential for equitable access.
Balanced perspective
Ultimately, successful clinical adoption will require harmonizing biological innovation with scalable manufacturing, regulatory clarity, and sustainable economic models to transition stem cell–derived β-cell therapies from experimental interventions to standard care for T1D.
Emerging technologies and future directions
Future advances in β-cell replacement therapy will rely on integrating computational, molecular, and bioengineering innovations to enhance graft durability and personalization. AI-driven graft monitoring using continuous glucose data, wearable biosensors, and imaging analytics may enable early detection of graft dysfunction and adaptive intervention. Digital phenotyping and multi-omics integration offer mechanistic insights into β-cell maturation, immune interactions, and predictive biomarkers of graft success. Concurrently, advances in genome engineering—combining HLA modulation with immune checkpoint and innate immune cloaking—alongside biomaterial-based vascularized scaffolds aim to achieve durable immune tolerance while minimizing reliance on systemic immunosuppression.
AI-driven graft monitoring and digital twins
AI and machine learning are increasingly integrated into β-cell replacement strategies to enable real-time graft monitoring and personalized care. AI-enabled biosensors, imaging analytics, and continuous glucose data can detect early graft dysfunction, while digital twin models allow individualized prediction of graft performance, immune compatibility, and therapeutic response, potentially reducing graft attrition9,80.
Multi-omics-guided donor matching and cell characterization
Multi-omics integration, encompassing genomics, transcriptomics, proteomics, and metabolomics, is transforming cell characterization and donor–recipient matching. These approaches identify molecular signatures associated with β-cell maturation, stress resilience, and immunogenicity, supporting optimization of differentiation protocols and standardization of allogeneic iPSC-derived products 10 .
Genome editing and immune-evasive cell platforms
Next-generation genome editing technologies, particularly CRISPR-Cas9-mediated knockout of HLA class I/II and overexpression of PD-L1 or CD47, are developing as effective methods to create “immune-invisible” β-cells. These genetic modifications reduce recognition by T cells and NK cells while maintaining metabolic function. Preliminary preclinical studies show enhanced graft survival without the need for systemic immunosuppression, representing a significant step forward in achieving functional immune tolerance. In addition, co-transplantation with MSCs provides further immunomodulation via paracrine signaling and the release of anti-inflammatory cytokines26,80.
Bioengineered vascularized scaffolds
Researchers are addressing graft hypoxia by developing vascularized scaffolds that incorporate pro-angiogenic or oxygen-releasing biomaterials such as VEGF-loaded hydrogels and microfluidic vascular templates. These bioengineered matrices enhance oxygen and nutrient diffusion, improve islet survival, and enable scalable manufacturing through 3D bioprinting or microfabrication 66 .
Novel perspectives
Emerging innovations are reshaping stem cell–derived β-cell therapy toward durable and personalized treatment for T1D. AI-driven graft monitoring, integrating continuous glucose monitoring and imaging analytics, enables early detection of graft dysfunction and supports individualized immunosuppression strategies 80 . Genome editing using CRISPR–Cas9 facilitates the generation of hypoimmunogenic β-cells through HLA modulation and immune-regulatory gene insertion, advancing universal donor concepts 8 . Novel transplantation niches, including liver-surface gelatin hydrogel scaffolds and prevascularized subcutaneous sites, improve oxygenation and engraftment 66 . In parallel, bioengineered encapsulation and oxygenation devices incorporating pro-angiogenic or oxygen-releasing biomaterials enhance graft survival and scalability 34 . Collectively, these advances may transition β-cell replacement from experimental therapy to a precision, potentially curative intervention.
Balanced interpretation: regulatory, cost, and scalability challenges
Despite significant advances in stem cell–derived β-cell therapy, its integration into clinical practice faces considerable translational challenges, particularly due to a complex regulatory environment. Stem cell interventions, classified as advanced therapy medicinal products (ATMPs), must comply with strict GMP requirements, long-term safety monitoring, and post-marketing surveillance. Furthermore, differing regulatory standards among agencies such as the FDA, EMA, and Central Drugs Standard Control Organisation (CDSCO) create varied approval processes, hindering global harmonization and implementation.
Scalability and standardization pose significant challenges in the generation of β-like cells, with varied differentiation protocols leading to inconsistencies in reproducibility, variability between batches, and quality control issues99,100. Although efforts are underway to develop large-scale bioreactor production platforms, their effectiveness in producing clinical-grade products reliably at a population scale has yet to be demonstrated 8 .
Long-term safety concerns in transplantation include risks of teratoma formation from residual undifferentiated cells, immune evasion potentially leading to malignancy, and off-target effects from genome-editing technologies. Addressing these issues necessitates longitudinal follow-up studies lasting over a decade.
Translating stem cell–derived β-cell therapy from research to clinical application requires overcoming regulatory, economic, manufacturing, and safety challenges to ensure the therapy is effective, safe, scalable, and accessible globally54,101,102.
Figure 3 presents a visual roadmap outlining the future directions of stem cell–derived β-cell therapy for T1D. It highlights key advancements such as genome editing for immune evasion, 3D organoid culture for improved maturation, AI-driven graft monitoring, and the development of personalized iPSC-based therapies. The roadmap emphasizes the integration of emerging technologies with translational and regulatory milestones, offering a forward-looking perspective on the pathway to clinical adoption.

Future roadmap of stem cell–based beta-cell therapy.
Conclusion
Stem cell–derived β-cell therapy represents a transformative advancement in the quest for a curative treatment for T1D. Over the past decade, rapid progress in differentiation protocols, immune modulation, and encapsulation technologies has moved the field closer to clinical realization. However, despite these achievements, persistent challenges such as immunogenicity, production cost, and regulatory standardization continue to constrain large-scale clinical implementation.
The translation of laboratory breakthroughs into clinically viable solutions will depend on strategic integration of stem cell biology, bioengineering, and computational innovation, including genome editing, AI-driven monitoring, and multi-omics-guided cell optimization. These multidisciplinary approaches are anticipated to enable durable, safe, and immune-tolerant islet regeneration, ultimately bridging the gap between experimental efficacy and long-term patient benefit.
In essence, the success of future β-cell replacement therapies will rely not merely on cellular differentiation but on creating a harmonized ecosystem that connects biological precision with clinical practicality, driving the next era of personalized regenerative medicine for diabetes care.
Footnotes
Acknowledgements
The author expresses sincere gratitude to Krescent Medical Research Pvt. Ltd. for their invaluable guidance and support throughout the preparation of this manuscript. Schematics were created with Canva and PowerPoint. Software tools used include BioRender.
Ethical considerations
Institutional Review Board approval is not required for this study.
Consent to participate
Patient consent is not required as there are no patients in this study.
Consent for publication
Not applicable.
Author contributions
All authors contributed substantially to the conceptualization, drafting, and critical revision of this manuscript. Dr Krishnaa S. Upadhye: Conceptualized the study, defined the design and intellectual content, conducted comprehensive literature searches, supervised clinical and experimental aspects, acquired relevant data, and took the lead in manuscript preparation, editing, and review. Dr Upadhye also served as the guarantor for the integrity and accuracy of the work.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Krishnaa S. Upadhye is the Director—Clinical Pharmacology & Drug Development at Krescent Medical Research Pvt. Ltd. This affiliation has been disclosed to the journal and the publisher. Krescent Medical Research Pvt. Ltd. had no role in the conceptualization, writing, or approval of this manuscript, and no funding was received from the company for this work. The views expressed in this review article are solely those of the author and do not necessarily reflect the views of Krescent Medical Research Pvt. Ltd.
Data availability statements
Not applicable, as no new data were generated or analyzed for this review article.
Statement of human and animal rights
This article does not contain any studies with human or animal subjects.
Statement of informed consent
There are no human subjects in this article and informed consent is not applicable.