
Abstract
Mitochondrial transplantation has emerged as a promising strategy for treating ischemic diseases by restoring mitochondrial function in damaged tissues. This study investigated the therapeutic potential of mitochondria isolated from placenta-derived mesenchymal stem cells (PD-MSCs) in a murine critical limb ischemia (CLI) model. The isolated mitochondria were characterized to confirm their structural integrity, purity, and ATP production capacity before transplantation into an ischemic hindlimb. Results showed that mitochondrial transplantation significantly improved blood flow and muscle regeneration compared with MSC transplantation, as evidenced by laser Doppler perfusion imaging and histological analysis. Enhanced ATP production and increased oxidative phosphorylation complex protein levels were observed, supporting energy metabolism in ischemic conditions. Mitochondrial transplantation also reduced mitochondrial reactive oxygen species (mROS) levels and increased antioxidant enzyme expression, including SOD-2, leading to reduced oxidative stress and apoptosis, as indicated by decreased Bax, cytosolic cytochrome c, and cleaved caspase-3 levels. Furthermore, mitochondrial transplantation promoted angiogenesis and increased vascular density in ischemic muscles by enhancing endothelial cell function. Overall, PD-MSC-derived mitochondrial transplantation demonstrated proved more effective over MSC transplantation in reducing inflammation, restoring mitochondrial function, and supporting tissue recovery, highlighting its promise as an effective therapeutic approach for CLI and other ischemic conditions by directly addressing mitochondrial dysfunction and overcoming the limitations of conventional cell therapies.

Introduction
Critical limb ischemia (CLI) is an advanced form of peripheral artery disease, characterized by chronic leg pain, paralysis, and tissue necrosis 1 . In its most severe form, CLI can lead to limb amputation or even death. Damage to the vessel walls, combined with ongoing cellular necrosis, leads to the formation of blood clots and their subsequent lysis, which results in chronic vascular obstruction2,3. As these clots continuously accumulate, they impede blood flow, ultimately cutting off the essential supply of oxygen and nutrients to the lower extremities and resulting in severe ischemia.
Given the severe consequences of CLI, effective therapeutic strategies should not only focus on restoring blood flow but also address the underlying ischemic damage at the cellular level. Current treatments for CLI, including surgical revascularization and pharmacological approaches, have focused on improving blood flow by removing blockages4,5. Therefore, a novel therapeutic approach is needed to mitigate ischemic injury, protect against oxidative stress, and promote neovascularization, ultimately improving patient outcomes.
Mitochondrial dysfunction plays a central role in CLI pathogenesis by driving oxidative stress and inflammation6,7. Ischemia-induced mitochondrial damage results in excessive reactive oxygen species (ROS) production, triggering lipid peroxidation and apoptosis, which further exacerbate tissue necrosis 8 . In addition, impaired mitochondrial bioenergetics compromises endothelial and muscle cell survival, limiting their ability to respond to ischemic stress and delaying tissue repair8–10. Given the critical role of mitochondria in cellular homeostasis, therapeutic approaches aimed at restoring mitochondrial function could offer significant advantages in treating CLI.
This study explores the potential of mitochondrial transplantation as a novel therapeutic strategy for CLI by directly targeting mitochondrial dysfunction. Mitochondria isolated from placenta-derived mesenchymal stem cells (PD-MSCs) were transplanted into ischemic hindlimbs to evaluate their effects on promoting neovascularization, reducing oxidative stress, and suppressing inflammation. Unlike conventional treatments that primarily focus on restoring vascular patency through mechanical or pharmacological means, mitochondrial transplantation offers a cell-free approach to modulating the ischemic microenvironment. By enhancing mitochondrial function, this strategy aims to improve tissue perfusion and vascular integrity, ultimately reducing disease progression and improving functional outcomes in CLI patients.
Materials and methods
Cell culture
This study was approved by the Institutional Review Board (IRB) of CHA University (IRB No. 201806-BR-29-02). PD-MSCs were obtained from CHA General Hospital, Seongnam, Korea. Cells were cultured in α-MEM (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone), 100 μg/ml streptomycin, 100 IU/ml penicillin, and 10 ng/ml basic fibroblast growth factor (CHA Meditech Co., Daejeon, Korea). Cells were subcultured at 80%–90% confluency and used at passage 7 for all experiments.
Mitochondria isolation
Mitochondria were isolated from PD-MSCs using a modified procedure. First, PD-MSCs were detached from T-175 flasks using 0.05% trypsin-EDTA and centrifuged at 1500 rpm for 5 min to obtain a pellet. The pellet was resuspended in 400 μl of SHE(+) buffer (0.25 M sucrose, 20 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.4, 2 mM EGTA (ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid), 10 mM KCl, 1.5 mM MgCl2, and 0.1% defatted BSA (bovine serum albumin)), supplemented with a protease inhibitor, and incubated on ice for 5 min.
Cells were lysed by passing them through a 26 G syringe 100 times on ice, after which 1.6 ml of SHE(+) buffer was added. The lysate was centrifuged at 1,000 × g for 5 min at 4°C to remove unbroken cells and debris. The supernatant containing mitochondria was then centrifuged at 12,000 × g for 10 min at 4°C to collect the mitochondria.
The isolated mitochondria were washed twice: first with SHE(−) buffer (0.25 M sucrose, 20 mM HEPES, pH 7.4, 2 mM EGTA, 10 mM KCl, 1.5 mM MgCl2) and then with Dulbecco’s phosphate buffered saline (DPBS). Finally, mitochondria were pelleted at 20,000 × g for 20 min at 4°C, resuspended in 200 μl of DPBS, and kept on ice for further experiments.
Transmission electron microscopy
Isolated mitochondria were fixed at 4°C in 2.5% glutaraldehyde solution containing 0.1 M phosphate buffer (pH 7.3) and post-fixed in 1% osmium tetroxide on ice for 2 h. After serial dehydration using ethanol and propylene oxide, the pellet was embedded in Epon 812. Ultrathin sections (70 nm) were prepared using an UltraCut UCT ultramicro-tome (Leica, Wien, Austria) and collected on 100-mesh copper grids. The sections were stained with 2% uranyl acetate and lead citrate and examined with a Tecnai G2 Spirit TWIN transmission electron microscope (FEI, OT, USA) at 120 kV.
To confirm the presence and localization of transplanted mitochondria within muscle tissue, skeletal muscle samples were harvested 7 days after mitochondrial transplantation. Tissues were fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) at 4 °C for 24 h. Post-fixation was performed with 1% osmium tetroxide for 1 h at room temperature. After dehydration in a graded ethanol series, samples were embedded in epoxy resin. Prior to transplantation, isolated mitochondria were labeled using 1.4 nm Nanogold Labeling Reagents (Nanoprobes, Yaphank, NY, USA; catalog #2025), according to the manufacturer’s instructions. Gold particles were covalently linked to thiol (-SH) groups present on the mitochondrial outer membrane via thiol-reactive chemistry, allowing direct and stable labeling without the use of antibodies. Tissue samples were processed using the same fixation, embedding, and staining methods described above. Ultrathin sections were stained with 2% uranyl acetate and lead citrate and examined under a Tecnai G2 Spirit TWIN transmission electron microscope (FEI) operated at 120 kV. Electron-dense Nanogold signals were observed within host muscle fibers, confirming the localization of transplanted mitochondria.
Murine model of hindlimb ischemia
Six- to 7-week old male Balb/c nude mice were obtained from Orient Bio (Seongnam, Korea) and housed in a pathogen-free facility at the Animal Center of CHA University under Specific Pathogen-Free (SPF) conditions. All animal procedures were approved by the Institutional Animal Care and Use Committee of CHA University (approval no. IACUC200133).
For the ischemia procedure, mice were anesthetized with 2% isoflurane, and an incision was made on the inner side of the right hindlimb to expose the superficial femoral artery. The artery was ligated at two points using triple surgical knots and incision was subsequently closed. Immediately after surgery, PD-MSCs (2 × 106 cells) and freshly isolated mitochondria (20 μg) were injected into four distinct intramuscular sites in the medial hindlimb, with each injection containing 10 μl (total volume of 40 μl per animal). Mitochondria were freshly isolated immediately prior to transplantation, and injections were administered 1 h after hindlimb ischemia (HLI) surgery to examine whether early mitochondrial delivery could support mitochondrial retention and mitigate acute ischemic injury. The experimental groups were (1) Normal (no surgery), (2) HLI (CLI surgery only), (3) HLI+MSC (CLI + PD-MSCs), (4) HLI+MT (CLI + mitochondria). Functional outcomes were evaluated at 7 days after transplantation, following euthanasia.
Perfusion imaging
Hindlimb blood flow was measured 7 days post-transplantation using a laser Doppler perfusion imager (LDPI; Moor Instruments, Devon, UK). The images were analyzed with MoorLDI Image Software (V5.3). Regions of interest (ROIs) were set around the legs, and perfusion ratios between ischemic and non-ischemic areas were calculated.
Measurement of ATP content
ATP content was measured using a modified CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega, Madison, WI, USA). To assess de novo mitochondrial ATP synthesis, 5 μl of 0.1 M ADP was added to 95 μl of culture medium as a substrate. This ADP-supplemented mixture was then combined with 100 μl of CellTiter-Glo reagent and applied to cells or mitochondria-containing preparations. The mixture was incubated for 45 min at room temperature to allow ATP generation, followed by 2 min of shaking. Luminescence was measured using a microplate reader (BioTek, Winooski, VT, USA). Net ATP production was calculated by subtracting the baseline luminescence (ADP-only control) from the total luminescence measured in the ADP-supplemented reaction.
MROS production
mROS in muscle tissue were assessed using MitoSOX (Molecular Probes; Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. Muscle tissues were frozen in optimal cutting temperature (OCT) compound and sectioned at a thickness of 10 μm using a cryostat. Sections were mounted on coated slides and incubated with 5-μM MitoSOX solution at 37°C for 30 min. After two PBS washes, the slides were visualized with a Zeiss 510 Confocal Laser Scanning Microscope. ROS production was quantified using Image J software.
Western blot analysis
Hindlimb tissue was homogenized in 1 ml of radio-immunoprecipitation assay (RIPA) lysis buffer (Biosesang, Seongnam, Korea), and proteins were collected by centrifugation at 12,000 rpm for 20 min at 4°C. Protein concentration was determined using a bicinchoninic acid (BCA) assay kit (Pierce; Thermo Scientific, Waltham, MA, USA). A total of 20 μg of protein was separated by electrophoresis on a polyacrylamide gel and transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Danvers, MA, USA).
The membrane was blocked with 5% BSA for 2 h and then incubated overnight at 4°C with primary antibodies, including anti-SOD-2 (1:1000; sc-137254, Santa Cruz, Dallas, TX, USA), Bcl-2 (1:1000; sc-23960, Santa Cruz), anti-Bax (1:1000; sc-7480, Santa Cruz), anti-Cytochrome c (1:1000; sc-13156, Santa Cruz), anti-β-actin (1:1000; sc-517582, Santa Cruz), anti-OXPHOS (1:1000; ab110413, Abcam, Cambridge, UK), anti-Caspase 3 (1:1000; ab4051, Abcam), anti-cleaved caspase 3 (1:1000; ab49822, Abcam), anti-VEGFA (1:1000; ab51745, Abcam), anti-VEGF-R2 (1:1000; ab39256, Abcam), anti-NF-kB p65 (1:1000; #8242, Cell Signaling, Danvers, MA, USA), and anti-phospho-NF-kB p65 (1:1000; #3033, Cell Signaling).
After overnight incubation, membranes were washed six times with Tris-buffered saline with Tween-20 (TBST) and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature. Protein bands were visualized using an ECL Plus kit (Bio-Rad, Hercules, CA, USA) and quantified using Image J software.
Histological staining analysis
Ischemic muscle tissue was embedded in OCT compound, frozen, and sectioned at 10 μm using a cryostat (Leica CM1950, Australia). Sections were stained with hematoxylin and eosin (H&E) and examined under an Olympus microscope.
Statistical analyses
Data are presented as mean ± SD. Statistical analyses were conducted using Sigma Plot 12.0 (Systat Software, San Jose, CA, USA). Group comparisons were made using the Mann–Whitney test and Student’s t-test. P-values less than 0.05 were considered statistically significant.
Results
Characterization of isolated mitochondria
PD-MSCs were pre-stained with MitoTracker Red CMXRos to evaluate mitochondrial viability, which depends on the mitochondrial membrane potential. The probe stains mitochondria based on their membrane potential, allowing visualization of viable mitochondria before and after isolation, without selectively distinguishing between different mitochondrial populations (Supplementary Fig. S1A). The reticulated morphology of isolated mitochondria was confirmed using transmission electron microscopy (TEM; Supplementary Fig. S1B). The identity of isolated mitochondria was verified by assessing functional markers, including VDAC, cytochrome c, and COX IV (Supplementary Fig. S1C). Mitochondrial activity was validated by measuring ATP content and synthesis. Isolated mitochondria, unlike damaged mitochondria (d-MT), generated ATP in response to ADP stimulation (Supplementary Fig. S1D). The presence of OXPHOS complex proteins in isolated mitochondria was also confirmed (Supplementary Fig. S1E).
Improvement of muscle function by transplanted mitochondria
Prior to inducing the CLI model, PD-MSCs were cultured and mitochondria were isolated. Mice were sacrificed at 7 days post-transplantation to analyze muscle recovery. The presence of transplanted mitochondria and cells was confirmed in the body up to 7 days post-transplantation (HLI+MT and HLI+MSC groups; Supplementary Fig. S2).
Blood perfusion, limb loss, foot necrosis, and limb salvage were monitored for 7 days following MT or MSC transplantation (Fig. 1b; n = 7 per group). The HLI group exhibited significant foot necrosis and limb loss due to ischemia. In contrast, both MT and MSC transplantation groups had reduced rates of foot necrosis and limb loss, with increased limb salvage. Notably, mitochondrial transplantation resulted in a threefold higher limb salvage rate compared with MSC transplantation (Fig. 1b).
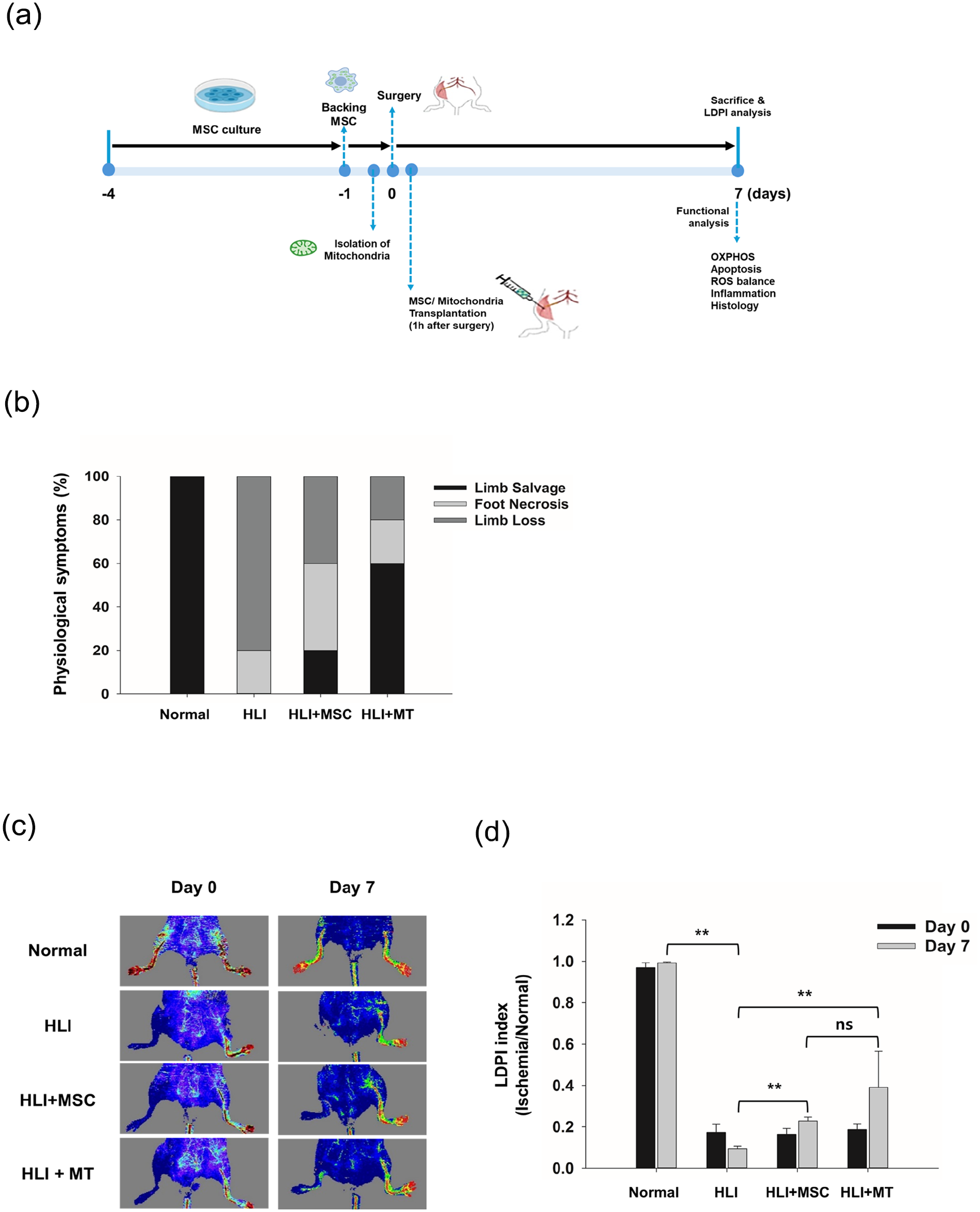
Therapeutic effects of mitochondrial transplantation in the hindlimb ischemia (HLI) model. (a) This diagram summarizes the HLI model experiment, showing the induction of ischemic injury and the timeline for analysis involving cell or mitochondrial transplantation. (b) Limb loss, foot necrosis, and limb salvage (absence of limb loss or foot necrosis) were quantified at day 7 post-femoral artery ligation following treatment with either MT or MSC transplantation (n = 7). (c) Representative laser Doppler perfusion imaging (LDPI) of the hindlimb in Balb/c nude mice, showing perfusion before and at day 7 after transplantation of isolated mitochondria or MSCs. (d) Quantification of hindlimb perfusion in Balb/c nude mice before and at day 7 after transplantation.
Blood reperfusion in the ischemic region was assessed using laser Doppler perfusion imaging at baseline and 7 days post-transplantation. The HLI group showed a significant reduction in perfusion compared with the non-ischemic side. These observations remained consistent over the 7-day period (Fig. 1c, d). In contrast, the MT and MSC transplantation groups demonstrated significant improvements in blood perfusion, with the MT group showing the most pronounced effect (Fig. 1c, d). These findings suggest that mitochondrial transplantation can effectively preserve ischemic tissue integrity and help preserve blood reperfusion capacity.
Regeneration of hindlimb muscle following mitochondrial transplantation
Changes in muscle mass were observed following mitochondrial transplantation (Fig. 2). The HLI group showed minimal increase in muscle mass due to ischemia. In contrast, mitochondrial transplantation resulted in a 30% increase in muscle mass, whereas MSC transplantation did not lead to significant recovery.

Therapeutic effects on muscle regeneration in the HLI model after mitochondrial transplantation. (a) Changes in skeletal muscle mass in each group at day 7 post-transplantation. (b) Representative H&E staining images showing changes in muscle fibers at day 7 after transplantation. (c) Transmission electron microscopy showing differences in skeletal muscle mitochondria between the groups at day 7 post-transplantation. (d) Quantification of mitochondrial content expressed as the number of mitochondria per field. (e, f) Electron microscopic evaluation of transplanted mitochondria using magnetic labeling.
Histological analysis was conducted to characterize muscle changes (Fig. 2b). H&E staining of ischemic muscle tissue in the HLI group revealed highly fragmented muscle fibers and tissue loss, indicating impaired function. In contrast, both the HLI+MT and HLI+MSC groups showed improved muscle fiber morphology and endomysium structure compared with the HLI group.
Similar trends were observed in TEM images, where the HLI group displayed compromised mitochondrial morphology, including structural damage to cristae (Fig. 2c). In contrast, mitochondrial transplantation effectively restored muscle cross-section morphology and mitochondrial structure, whereas MSC transplantation showed limited improvement.
Mitochondrial quantification in ischemic muscle 7 days post-transplantation further supported these findings (Fig. 2d). Although muscle loss remained significant reduction compared with the normal group, mitochondrial transplantation led to a 2.4-fold increase in mitochondrial content relative to the untreated HLI group, suggesting partial restoration of mitochondrial content rather than complete muscle regeneration. No significant changes were observed in the MSC group.
To confirm that these changes were specifically due to transplanted mitochondria, magnetically labeled mitochondria were tracked (Fig. 2e, f). The labeled mitochondria were found distributed among muscle fibers, supporting their role in muscle regeneration. Overall, these results suggest that transplanted mitochondria contribute significantly to the regeneration of damaged muscle.
Restoration of mitochondrial function in muscle by transplanted mitochondria
To evaluate the effect of mitochondrial transplantation on mitochondrial function in ischemic muscles, we analyzed the expression of OXPHOS complex proteins (Fig. 3). The HLI group showed a decrease in OXPHOS protein levels compared with the normal group (Fig. 3a, b). However, the HLI+MT group demonstrated an increase in OXPHOS protein expression. No significant changes in OXPHOS protein levels were observed in the HLI+MSC group (Fig. 3a, b).

Therapeutic effects on muscle mitochondrial function in the HLI model. (a, c) Immunoblot analysis of mitochondrial OXPHOS complexes (a) and COX IV (c) in each group at day 7 after transplantation. (b, d) Quantification of OXPHOS complex and COX IV expression for panels A and C, respectively, normalized to total protein. (e) Measurement of ATP content in each group at day 7 post-transplantation using the CellTiter-Glo luminescence assay. Data are presented as mean ± SEM, *P < 0.05; **P < 0.01; ***P < 0.001.
Similarly, COX IV expression and ATP content showed the same trend (Fig. 3c–e). These findings suggest that transplanted mitochondria restore mitochondrial function impaired by ischemic damage, leading to increased ATP production in the muscle.
Regulation of apoptosis through the balance of ROS production and antioxidant defense by transplanted mitochondria
To investigate the molecular mechanisms of limb ischemic pathogenesis, we examined the balance between ROS production and antioxidant defense following early ischemic injury. The HLI group showed increased mROS and decreased SOD-2 expression compared with the normal group, indicating the effects of ischemic injury (Supplementary Fig. S3). Conversely, both the MSC and mitochondrial transplantation groups showed changes in mROS production and SOD expression (Supplementary Fig. S3A). Notably, mitochondrial transplantation was more effective in suppressing mROS production compared with MSC transplantation (57.1% vs. 21.4%; in Supplementary Fig. S3A). SOD-2 expression in the mitochondrial transplantation group was restored to normal levels (Fig. 3b).
Altered antioxidant defense mechanisms due to ischemia can lead to apoptosis 7 . To verify this, we examined the protein levels of apoptotic markers, including Bax, cytosolic cytochrome c, and cleaved caspase-3 (Fig. 4a, b). The HLI group showed increased levels of Bax, cytosolic cytochrome c, and cleaved caspase-3 compared with the normal group (Fig. 4a, b). In contrast, the mitochondrial transplantation group showed a marked reduction in all apoptotic markers. The MSC group showed about a 50% reduction in Bax and cytosolic cytochrome c, but no significant effect on cleaved caspase-3 expression (Fig. 4a, b). In addition, the anti-apoptotic protein BCL2 was elevated in the HLI group compared with the normal group, likely reflecting a compensatory cellular response to ischemic stress. Although BCL2 levels remained moderately increased in the transplantation groups, the BCL2/BAX ratio was lower in the HLI+MT group, which may suggest a shift toward restored apoptotic balance rather than increased pro-apoptotic signaling. These results suggest that transplanted mitochondria can regulate apoptosis by balancing ROS production and antioxidant defenses.

Exogenous mitochondria regulate apoptosis-related proteins via the NF-κB pathway in CLI mice. (a) Immunoblot analysis of apoptosis-related markers (Bcl-2, BAX, cytosolic cytochrome c, caspase-3, and cleaved caspase-3) in each group at day 7 post-transplantation. (b) Quantification of the results for panel A. Data are presented as mean ± SEM, *P < 0.05; **P < 0.01; ***P < 0.001.
Promote angiogenesis and alleviate inflammatory response by transplanted mitochondria
ROS stimulates pro-inflammatory cytokine release by activating NF-κB signaling pathways 11 . To investigate this, we analyzed changes in NF-κB expression (Fig. 5a, b). The HLI group showed increased NF-κB activation, whereas the mitochondrial transplantation group demonstrated decreased NF-κB expression. No significant changes were observed in the MSC transplantation group (Fig. 5a, b).

Therapeutic effects on inflammation and angiogenesis markers in the HLI model. (a) Immunoblot analysis of NF-κB phosphorylation across groups at day 7 post-transplantation. (b) Quantification of NF-κB phosphorylation shown in panel A. (c) Immunoblot analysis of VEGF-A and VEGF-R2 across groups at day 7 post-transplantation. (d) Quantification of VEGF-A and VEGF-R2 expressions shown in panel C. Data are presented as mean ± SEM, *P < 0.05; **P < 0.01; ***P < 0.001.
To determine whether transplanted mitochondria stimulate angiogenesis, we examined the expression of angiogenic growth factors, VEGF and VEGFR-2, in the experimental groups (Fig. 5c, d). The HLI group showed a significant decrease in VEGFR-2 expression compared with the normal group. In contrast, the mitochondrial transplantation group showed a twofold increase in VEGFR-2 expression. No significant changes were observed in the MSC transplantation group (Fig. 5c, d).
Overall, these results indicate that transplanted mitochondria can inhibit inflammation and promote angiogenesis.
Discussion
In this study, we demonstrated the therapeutic potential of PD-MSC-derived mitochondria for treating CLI in a murine model. Our findings show that mitochondrial transplantation not only enhanced blood flow (Fig. 1) and muscle recovery (Fig. 2) but also restored mitochondrial function (Fig. 3), reduced apoptosis (Fig. 4), and minimized oxidative stress (Supplementary Fig. S3). These therapeutic outcomes were superior to those observed with MSC transplantation alone, highlighting the ability of mitochondrial transplantation to overcome the limitations of traditional stem cell therapies by directly addressing mitochondrial dysfunction, a key contributor in ischemic damage. In addition, these results suggest a broader applicability for treating other mitochondrial dysfunction–related diseases, aligning with previous reports7,8,12.
The characterization of isolated mitochondria confirmed their structural and functional viability (Supplementary Fig. S1), which laid a solid foundation for subsequent transplantation experiments aimed at tissue repair. The ability of these mitochondria to maintain structural integrity and generate ATP (Supplementary Fig. S1D, E) was crucial in supporting their functional role in tissue regeneration. This initial characterization ensured that the transplanted mitochondria were capable of restoring physiological function in the ischemic environment.
Mitochondria’s role in oxidative stress reduction and apoptosis suppression is well-documented in ischemic models8,13. Our study adds further evidence to these findings by demonstrating that mitochondrial transplantation effectively reduces oxidative stress (Supplementary Fig. S3) and prevents cell death in ischemic muscle (Fig. 4). Specifically, the reduction in apoptotic markers, such as Bax, cytosolic cytochrome c, and cleaved caspase-3, suggests that transplanted mitochondria actively protect cells from ischemic injury. This effect is partly due to the enhancement of antioxidant defenses, as evidenced by the upregulation of SOD-2 expression in the mitochondria-treated group (Supplementary Fig. S3B). Restoring antioxidant balance directly targets central regulators of apoptosis, mitigating the damaging feedback loop caused by excessive ROS production.
While BCL2 is generally known for its anti-apoptotic function, its upregulation in the HLI group may represent a stress-induced compensatory mechanism rather than effective protection. Notably, although BCL2 levels remained elevated, the lower BCL2/BAX ratio in the HLI+MT group suggests that mitochondrial transplantation may normalize apoptotic signaling, potentially by alleviating cellular stress and modulating both pro- and anti-apoptotic pathways.
Our results also showed improvements in ATP production and OXPHOS complex activity (Fig. 3), which are crucial for muscle regeneration in ischemic conditions. The enhanced bioenergetics provided by mitochondrial transplantation supports increased cellular energy demands, which is essential for effective tissue repair and regeneration13–15.
While the observed increases in ATP content and OXPHOS protein levels may partially reflect the preservation of host muscle cells, we cautiously propose that these changes also indicate a direct bioenergetic contribution from the transplanted mitochondria. Previous reports have demonstrated that isolated mitochondria can produce ATP independently of nuclear transcription and exert metabolic effects shortly after delivery, particularly during the acute phase of ischemic injury. For instance, previous studies have reported that transplanted mitochondria increase local ATP levels and reduce tissue damage in ischemic heart and spinal cord models, providing additional support for the potential bioenergetic contribution of transplanted mitochondria 16 . However, it remains unclear whether the observed mitochondrial signal truly represents successful integration of exogenous mitochondria into host cells. Since skeletal muscle naturally has high mitochondrial density, the apparent increase may result from preserved tissue rather than incorporation. Validation through species-specific mtDNA quantification or dual labeling is needed. In addition, the observed ATP increase might be attributed to reduced muscle damage and cell death, rather than a direct enhancement of mitochondrial function. Further studies are essential to clearly distinguish between metabolic rescue and tissue preservation mechanisms. Thus, we interpret our findings as a combined result of reduced tissue loss and early metabolic support by the exogenous mitochondria.
Previous studies have demonstrated that mitochondrial transplantation not only improves cellular metabolism but also reduces oxidative stress and apoptosis in various ischemic tissue models, including cardiac ischemia 17 . These findings emphasize the importance of targeting mitochondrial bioenergetics as a therapeutic approach for enhancing recovery in ischemic tissues. Compared with previous studies on MSC transplantation for CLI or other ischemic diseases, our data potentially suggest that mitochondrial transplantation provides more direct and rapid support of mitochondrial function. However, our findings should be interpreted with caution, as the limited set of mechanistic data do not definitively establish mechanisms such as angiogenesis or muscle regeneration. Notably, previous work has shown that mitochondria delivered within a short window after ischemic onset can preserve tissue function and reduce oxidative injury, supporting the rationale for early-phase transplantation 16 . Further investigation is essential to delineate the molecular mechanisms and long-term outcomes associated with mitochondrial therapy.
The ability of transplanted mitochondria to reduce oxidative stress is pivotal for protecting cells from ischemic damage. Excessive ROS production in ischemic tissues exacerbates injury by promoting inflammatory signaling and cellular apoptosis 8 . In our study, mitochondrial transplantation resulted in a marked reduction in mROS levels (Supplementary Fig. S3A) and restoration of antioxidant enzymes SOD-2 (Supplementary Fig. S3B), which are key mediators of cellular redox balance. Although these outcomes suggest a protective role of the transplanted mitochondria, the underlying mechanism remains to be clarified. It is not yet clear whether this effect is mediated by direct mitochondrial incorporation into host cells and their functional integration, or through indirect mechanisms such as paracrine signaling or the enhancement of endogenous antioxidant responses. Future studies employing co-localization analysis, mitochondrial transfer tracking, and real-time ROS imaging are critical to delineate the precise pathways involved. In addition, while we observed reduced NF-κB expression following mitochondrial transplantation, the absence of histological validation—such as immunostaining for infiltrating immune cells—limits our ability to interpret this as definitive evidence of inflammation suppression. Further investigation using cell-type-specific markers (e.g., CD68 for macrophages, MPO for neutrophils) would help clarify whether mitochondrial transplantation directly modulates immune cell infiltration and inflammatory status at the tissue level. These findings align with previous reports demonstrating that mitochondrial transplantation can attenuate oxidative stress and improve tissue outcomes in ischemia-reperfusion models, including in a porcine cardiac setting, although the underlying mechanisms remain to be elucidated 18 . By restoring antioxidant defenses, mitochondrial transplantation may interrupt the cycle of oxidative injury and apoptosis, thereby enhancing tissue viability in ischemic conditions.
Another significant observation in our study was the promotion of angiogenesis following mitochondrial transplantation (Fig. 5b). Increased vascular density in ischemic muscles, as supported by histological analysis, suggests that transplanted may enhance blood supply by supporting endothelial cell metabolism and mitochondrial function. Angiogenesis plays a vital role in tissue recovery by ensuring oxygen and nutrient delivery to regenerating regions. Previous studies have emphasized the importance of mitochondrial health in endothelial function and angiogenic capacity 19 .
However, we acknowledge that VEGFR2 expression alone is insufficient to confirm angiogenesis. Direct quantification of neovascularization using endothelial markers such as CD31 or isolectin B4 would be required to validate this observation. In addition, although VEGF-A and VEGFR2 expression were elevated following transplantation, these changes future studies are necessary to incorporate immunohistochemical markers such as CD31 or isolectin B4 to accurately quantify neovascularization. VEGF signaling can be activated in response to ischemic stress or paracrine signaling and does not necessarily indicate direct involvement of viable transplanted cells. Moreover, whether transplanted mitochondria directly stimulate endothelial cells to initiate angiogenic programs requires mechanistic investigation using functional and cell-type-specific assays, as functional assays and cell-type-specific analyses were not conducted in this study. In particular, in the +MSC group, the modest VEGF expression may not reflect sustained angiogenic activity due to the likely lack of MSC engraftment or limited secretory function.
Likewise, the observed preservation of muscle architecture may be the result of tissue protection rather than de novo regeneration. Without specific markers of myogenesis or structural evidence of new fiber formation, the regenerative effect of mitochondrial transplantation cannot be definitively concluded. These considerations highlight the need for further studies combining functional, molecular, and histological endpoints to comprehensively assess both vascular remodeling and muscle recovery after mitochondrial transplantation.
Despite the promising results, several challenges remain for the clinical translation of mitochondrial transplantation. Although mitochondria exhibit low immunogenicity, further studies are needed to evaluate their long-term safety and potential immune responses in human subjects. Moreover, the optimal timing of transplantation remains uncertain. While we administered mitochondria 1 h post-injury to examine early intervention effects, it is possible that the acute inflammatory environment may have impacted mitochondrial retention and function. Comparing early versus delayed transplantation (e.g., 24 h) would help define the most effective therapeutic window. In contrast, the therapeutic effect of MSC transplantation was limited, which may reflect poor cell survival after injection. We did not perform engraftment tracking in this study, and future experiments incorporating cell labeling strategies would be necessary to evaluate MSC persistence. In addition, the relatively short follow-up duration (2 weeks) may not fully capture the long-term effects of mitochondrial transplantation on functional recovery and vascular remodeling, potentially underestimating the durability of therapeutic benefits. Extending the observation period to 4–8 weeks should be incorporated in future studies to more comprehensively assess the durability and long-term outcomes of mitochondrial transplantation. Finally, the development of standardized dosing strategies and delivery methods remains essential to ensure the scalability of mitochondrial therapies. Future research should focus on refining these parameters to enable the successful clinical application of mitochondrial transplantation for CLI and other ischemic conditions.
In summary, our findings provide compelling evidence that mitochondrial transplantation is a promising therapeutic strategy for CLI. By targeting mitochondrial dysfunction—a key driver of ischemic tissue injury—this approach offers advantages beyond traditional cell-based therapies. Mitochondrial transplantation not only improved tissue perfusion and preserved muscle structure but also restored mitochondrial function, reduced oxidative stress, and attenuated apoptosis. Nevertheless, this strategy is still in the early stages of development, and several important questions remain. Limitations regarding optimal timing, delivery methods, cell-free safety, and long-term efficacy must be addressed. Future studies should focus on refining transplantation protocols, identifying reliable efficacy markers, and extending the evaluation period to assess the durability of therapeutic effects. Taken together, these findings support the continued exploration of stem cell–derived mitochondria as a scalable and targeted treatment modality for ischemic diseases.
Supplemental Material
sj-docx-1-cll-10.1177_09636897251347391 – Supplemental material for Comparative analysis of mitochondrial and mesenchymal stem cell transplantation for angiogenesis and muscle regeneration
Supplemental material, sj-docx-1-cll-10.1177_09636897251347391 for Comparative analysis of mitochondrial and mesenchymal stem cell transplantation for angiogenesis and muscle regeneration by Mi Jin Kim, Jung Wook Hwang, Chang-Koo Yun, Ikhyun Lim, Kyunghoon Min and Yong-Soo Choi in Cell Transplantation
Footnotes
Ethical Considerations
This study was approved by our Institutional Review Board (IRB) of CHA University (IRB No. 201806-BR-29-02). All animal procedures were approved by the Institutional Animal Care and Use Committee (approval No. IACUC200133) of CHA University on September 1, 2020.
Author Contributions
Y.-S.C. supervised the project and conceived the idea of mitochondrial transplantation for CLI; J.W.H. performed the experiments; M.J.K. and Y.-S.C. wrote the paper. M.J.K., J.W.H., C.-K.Y., I.L., K.M. and Y.-S.C. reviewed and approved the final version of this manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Bio and Medical Technology Development Program of the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT (grant no. NRF-2018M3A9B5023052) and by a grant of Korean Fund for Regenerative Medicine (KFRM) grant funded by the Korea government (the Ministry of Science and ICT, the Ministry of Health & Welfare) (grant no. 22C0609L1).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Data Availability Statement
All data are available from the corresponding author on request. Most relevant data are already included.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.