
Abstract
Pancreatic islet transplantation (PITx) is a promising treatment option for patients with type 1 diabetes mellitus. Previously, we demonstrated that therapy with alloantigen-specific immunomodulatory cells (IMCs) generated ex vivo in the presence of anti-CD80 and CD86 monoclonal antibodies (mAbs), successfully induced tolerance following clinical liver transplantation. To extend IMC therapy to PITx, it is crucial to address the strong inflammatory and innate immune responses that occur immediately after PITx. In this study, we investigated the efficacy of IMCs in modulating macrophage activation and mitigating inflammatory damage of pancreatic islets. IMCs were induced using mouse splenocytes in the presence of anti-mouse anti-CD80 (RM80) and anti-CD86 (GL-1) mAbs. IMCs exerted donor-specific immunosuppressive effects in a mixed lymphocyte reaction. During lipopolysaccharide (LPS) stimulation, the addition of IMCs suppressed conversion to the M1 phenotype and promoted a shift toward the M2 phenotype, particularly under direct cell–cell contact conditions. Nitric oxide production, a hallmark of M1 polarized macrophages, was significantly reduced in LPS-stimulated RAW264 macrophages by IMC treatment. These findings were associated with reduced secretion of pro-inflammatory cytokines, tumoral necrosis factor α, and interleukin-6, and increased interleukin-10 production by macrophages. IMCs effectively prevented macrophage-mediated islet destruction after 12 h of co-culture with LPS-stimulated macrophages and significantly inhibited macrophage migration toward allogeneic islets in vitro. Intraportal co-infusion of IMCs with syngeneic islets in a mouse PITx model resulted in reduced messenger RNA (mRNA) expression of pro-inflammatory cytokines in the recipient liver. Immunohistochemical staining revealed a significantly lower number of F4/80+ macrophages at the transplantation site in IMCs-treated mice. These results demonstrate that IMCs modulate macrophage polarization, promoting a shift toward the M2 phenotype and protecting islets from macrophage-mediated damage. These effects combined with its intrinsic donor antigen-specific immunosuppressive capacity make IMC therapy a promising strategy for improving outcomes after PITx.

Keywords
Introduction
Pancreatic islet transplantation (PITx) is an effective treatment option for patients with type 1 diabetes mellitus with hypoglycemia unawareness or severe hypoglycemic events 1 . This minimally invasive procedure involving the infusion of islets into the portal vein enables β-cell replacement, offering the potential for a cure for diabetes. The rate of insulin independence after PITx has increased substantially in recent years owing to refinements in islet isolation techniques2,3 and the groundbreaking protocol introduced by the Edmonton group in 20004–6. According to the latest report by the Collaborative Islet Transplant Registry in 2022, approximately 90% of patients achieved abrogation of severe hypoglycemic events, 60% of patients maintained an HbA1c level of <7.0%, and 30% of patients achieved an insulin-independent state at 5 years of follow-up after PITx 7 . Although not all transplant recipients achieve insulin independence, even minimal graft function can protect against hypoglycemia 8 , improve quality of life 9 , and prevent the progression of microangiopathy 10 . In addition, previous reports have shown that PITx provides better glycemic control than optimal medical therapy11–13, which was further supported by the first randomized controlled clinical trial comparing PITx with insulin therapy 14 .
However, several challenges must be addressed before this procedure can become a mainstream strategy for treating insulin deficiency. The current need for lifelong immunosuppression, which has numerous adverse effects, represents a significant challenge that compromises transplantation outcomes. The increased risk of opportunistic infections, cardiovascular diseases, malignancies, renal insufficiency, and diabetes can severely affect patients’ quality of life15,16. Moreover, existing immunosuppression protocols often fail to ensure long-term islet acceptance, and islet rejection remains a significant concern after PITx. Inducing specific immune tolerance against donor antigens has emerged as the ultimate goal in transplantation medicine, as it can prevent allograft rejection while avoiding the side effects associated with non-specific immunosuppression. Over the past few decades, several approaches have been explored to achieve tolerance; however, the most effective method for clinical application in humans remains unclear. Cell-based strategies, such as the adoptive transfer of regulatory cells, show particular promise for inducing tolerance after transplantation17,18. Building on previous findings from a non-human primate kidney transplant model 19 , we have previously evaluated the safety and efficacy of cell therapy in the context of living-donor liver transplantation 20 . In this study, immunomodulatory cells (IMCs) were generated through a 2-week co-culture of recipient peripheral blood mononuclear cells (PBMCs) with irradiated donor PBMCs in the presence of anti-CD80 and anti-CD86 monoclonal antibodies (mAbs). Remarkably, immunosuppression was successfully discontinued in 7 of 10 recipients, achieving stable tolerance with no histopathological signs of rejection for over 10 years 21 . This exciting and promising outcome led to the idea of applying IMC therapy in patients receiving PITx.
Unlike in the case of solid organ transplants, the primary non-function of transplanted islets is driven by factors other than the cell-mediated immune response. The innate immune response combined with islet isolation impairs islet engraftment, leading to substantial early islet graft loss 22 . Once isolated islets are infused into the recipient’s circulatory system, they induce platelet aggregation and activate the coagulation and complement cascades 23 . This triggers a strong inflammatory response, resulting in the production of pro-inflammatory cytokines, such as interferon (IFN)-γ, interleukin (IL)-2, IL-12, tumoral necrosis factor (TNF)-α, monocyte chemoattractant protein-1 (MCP-1), and macrophage inflammatory protein (MIP)-1β. These cytokines rapidly recruit macrophages, neutrophils, and cytotoxic leukocytes to the transplantation site, directly injuring the transplanted islets and amplifying the inflammatory response23,24. Macrophages, the key effector cells of the innate immune system, prime adaptive immune responses and promote islet graft loss following PITx 25 . Transient macrophage inhibition and/or depletion has been shown to significantly prolong pancreatic islet graft survival 26 , highlighting the importance of ameliorating inflammatory responses after PITx to improve graft survival27,28. These inflammatory responses not only affect allogeneic or xenogeneic islet transplants, but autologous islets are also subjected to innate inflammatory damage.
In this study, we aimed to evaluate the efficacy of IMC treatment in improving the outcomes after PITx. We explored the anti-inflammatory potential of IMCs to modulate the activation of innate immune cells, with the objective of addressing the specific challenge of inflammation-mediated islet damage following PITx.
Materials and Methods
Animals
Male C57BL/6 (H-2b), BALB/c (H-2d), and C3H/HeJ (H-2k) mice were purchased from SLC (Shizuoka, Japan). The mice were maintained in a specific pathogen-free facility and used for the experiments at 12 to 15 weeks of age. The C57BL/6-Ly5.1 mouse strain (RBRC00144) was provided by RIKEN BRC through the National BioResource Project of the MEXT/AMED, Japan. All experimental procedures were conducted in strict accordance with the Guidelines for the Care and Use of Laboratory Animals of Hokkaido University Graduate School of Medicine and approved by the Institutional Animal Care Committee (approval no. 22-0048).
Reagents
Anti-mouse anti-CD80 (RM80) and anti-CD86 (GL-1) mAbs were provided by Juntendo University. Lipopolysaccharide (LPS, Escherichia coli:055: B5) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Collagenase P was purchased from Roche (Basel, Switzerland). A nitrite colorimetric assay was performed using the Griess reagent (Sigma-Aldrich).
Induction of IMCs
The IMCs were generated based on the method previously described20,29. Briefly, splenocytes from C57BL/6 mice (40 × 106) were co-cultured with irradiated BALB/c splenocytes (30-Gy, 40 × 106) in the presence of anti-CD80 and CD86 mAbs (10 μg/mL each) in Roswell Park Memorial Institute (RPMI)-1640 medium (Sigma-Aldrich) supplemented with 10% heat-inactivated fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin (Sigma-Aldrich) at 37°C with 5% CO2 in a humidified atmosphere. After 7 days of the co-culture, viable cells were collected and used as IMCs for further analyses. Splenocytes cultured without anti-CD80 and anti-CD86 antibodies were also assessed (Fig. 1).

Immunomodulatory cell induction. Diagram showing the protocol used to induce murine IMCs. C57BL/6 splenocytes are stimulated with irradiated BALB/c splenocytes in the presence of anti-mouse anti-CD80/86 mAbs for 7 days. aCD80/aCD86 mAbs: anti-mouse anti-CD80 and anti-CD86 monoclonal antibodies; IMCs: immunomodulatory cells.
Macrophage Preparation and Culture
The RAW264 cells (derived from BALB/c mice, H-2d) were obtained from Riken BioResource Center (Tsukuba, Japan). The cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Nacalai Tesque, Kyoto, Japan) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin.
Flow Cytometry
IMCs and control cells cultured without anti-CD80/86 mAbs were stained with fluorochrome-conjugated antibodies against various lineage cell surface markers. The following antibodies were used: PerCP-conjugated 7AAD (BD Biosciences, cat# 559925), FITC-conjugated CD11c (BD Biosciences, cat# 553801), PE-conjugated CD11b (BD Biosciences, cat# 557397), PE-Cy7-conjugated CD3e (BD Biosciences, cat# 552774), APC-conjugated FOXP3 (Invitrogen, cat# 17-5773-82), APC-conjugated Ly-6G and Ly-6C (BD Biosciences, cat# 561083), APC-Cy7-conjugated NK-1.1 (BD Biosciences, cat# 560618), APC/Fire 750-conjugated CD19 (BioLegend, cat# 115557), APC-H7-conjugated CD4 (BD Biosciences, cat# 560181), eF450-conjugated anti-F4/80 (Invitrogen, cat# 48-4801-82), BV421-conjugated CD25 (BD Biosciences, cat# 562606), and eF506-conjugated CD8a (Invitrogen, cat# 69-0081-82). After staining, the cells were washed with phosphate-buffered saline (PBS) to remove excess antibodies. A minimum of 100,000 events were acquired using a BD FACS Canto II system (BD Biosciences, San Jose, CA, USA).
For the M1/M2 macrophage polarization assessment, the collected RAW264 macrophages were incubated with fluorochrome-labeled antibodies against M1 and M2 macrophage surface markers for 30 min at 4°C. The following antibodies were used: PerCP-conjugated 7AAD (BD Biosciences, cat# 559925), PE-conjugated anti-CD206 (BD Biosciences, cat# 568273), PE-Cy7-conjugated anti-CD86 (BioLegend, cat# 105013), APC-conjugated anti-CD163 (BioLegend, cat# 155305), APC/Fire 750-conjugated anti-CD80 (BioLegend, cat# 104739), and eF450-conjugated anti-F4/80 (Invitrogen, cat# 48-4801-82). After staining, the cells were washed with PBS to remove excess antibodies. A minimum of 100,000 events were acquired using a BD FACS Canto II system.
Mixed Lymphocyte Reaction
Cell proliferation was tracked using the fluorescent dye, CellTrace Violet (Thermo Fisher Scientific, Philadelphia, PA, USA), as previously described30,31. Splenocytes from C57BL/6-Ly5.1 congenic mice were used as responder cells, while 30-Gy irradiated BALB/c splenocytes served as stimulator cells. Responder cells were stained with CellTrace Violet and co-cultured in 96-well round-bottom plates with the stimulator or third-party cells (C3H/HeJ, H-2k), with or without varying numbers of IMCs (5.0 × 104 to 6.25 × 103/well). After 6 days of culture, the percentage of dividing T cells labeled with CellTrace Violet was evaluated by flow cytometry using the 405-450/50 nm detection channel.
Measurement of Nitric Oxide Production
Nitric oxide (NO) synthesis was assessed by quantifying the concentration of nitrite, a stable metabolite of NO, in the culture supernatants using the Griess assay, as previously described 32 . Briefly, RAW264 macrophages were seeded in 24-well plates and co-cultured with or without IMCs. After stimulation with LPS for 48 h, culture supernatants were mixed with an equal volume of Griess reagent (100 µL; Sigma-Aldrich) in 96-well plates and incubated at room temperature for 10 min, following the manufacturer’s instructions. Subsequently, the absorbance was measured at 540 nm using a microplate reader (Varioskan; Thermo Fisher Scientific), and the nitrite concentration was calculated using a nitrite standard curve.
Effects of IMCs on Macrophage Activation In Vitro
IMCs were co-cultured with RAW264 macrophages using a cell–cell contact or separate co-culture model. In the direct cell–cell contact co-culture model, RAW264 macrophages were seeded in 12-well plates (5.0 × 105 cells/well) and treated with IMCs (1.0 × 106 cells). LPS was added to the culture media to a final concentration of 100 ng/mL, and cells were incubated at 37°C with 5% CO2 in a humidified atmosphere for 6 h. Macrophages were collected using ice-cold PBS with 5 mM ethylenediaminetetraacetic acid (EDTA) for flow cytometry, and supernatants were collected for cytokine analysis.
In the separate co-culture model, RAW264 macrophages were seeded in the lower chamber (5.0 × 105 cells/well), while IMCs (1.0 × 106 cells) were seeded on inserts with a 0.4-µm pore size, sufficient to prevent cell passage through the membrane (Corning, NY, USA, cat. 3401). LPS was added to the medium to a final concentration of 100 ng/mL. As in the cell–cell contact co-culture model, macrophages and supernatants were collected and used for further analysis.
Measurement of Cytokines
The TNF-α, IL-6, and IL-10 protein levels were quantified by enzyme-linked immunosorbent assay (ELISA) using ELISA Flex kits (Mabtech, Sweden) following the manufacturer’s instructions. All measurements were performed in duplicates.
Cytokine Gene Expression: Messenger RNA Extraction and Quantitative Polymerase Chain Reaction
The messenger RNA (mRNA) was extracted from the liver samples using the PureLink RNA Mini kit (Invitrogen, CA, USA), following the manufacturer’s protocol. Total mRNA concentration was measured using a NanoDrop 2000c spectrophotometer (Thermo Scientific). The complementary DNA (cDNA) was synthesized from 1 µg of complementary mRNA using a ReverTra Ace kit (Toyobo, Osaka, Japan). TB Green Premix Ex Taq II reagents (Takara, Shiga, Japan) were used for detection, and the expression of the target mRNA was determined using a StepOne Plus Real-Time PCR System (Applied Biosystems, Life Technologies). The following primers were used: mouse TNF-α forward 5ʹ-ACCCTCACACTCAGATCATC-3ʹ and reverse 5ʹ-GAGTAGACAAGGTACAACCC-3ʹ, mouse IL-1β forward 5ʹ-AAAGCTCTCCACCTCAATGG-3ʹ and reverse 5ʹ-AGGCCACAGGTATTTTGTCG-3,ʹ mouse MCP-1 forward 5ʹ-TCCCAATGAGTAGGCTGGAG-3ʹ and reverse 5ʹ-TCTGGACCCATT-CCTTCTTG-3ʹ, MIP-1β forward 5ʹ-CCCACTTCCTGCTGTTTCTC-3ʹ and reverse 5ʹ-GTCTGCCTCTTTTGGTCAGG-3ʹ, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward 5ʹ-TACACTGAGGACCAGGTTG-3ʹ and reverse 5ʹ-CTGTAGCCG-TATTCATTGTC-3ʹ. The mean cycle threshold (Ct) value of the housekeeping gene, GAPDH, was used to normalize target gene expression.
Pancreatic Islet Isolation
The pancreatic islets of C57BL/6 or BALB/c mice were isolated as previously described 27 . The isolated islets were cultured overnight in RPMI-1640 medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin, and 0.25 μg/mL amphotericin B (Sigma-Aldrich) at 37°C with 5% CO2 in a humidified atmosphere. Purified islets with diameters ranging from 150 to 200 μm were used for the experiments.
Islet-Macrophage Co-Culture Model
RAW264 macrophages were seeded into 12-well plates and stimulated with LPS (100 ng/mL). BALB/c islets were seeded on top of the macrophage monolayer (100 islets/well), and IMCs were added to the treated groups. After 12 h of co-culture, the number of pancreatic islets was assessed.
Macrophage Migration Assay
The effect of IMCs on macrophage migration toward allogeneic islets was evaluated using a separate co-culture model (Corning, cat. 3421), as previously described 33 . IMCs were seeded in the lower wells of a 24-well Transwell plate, in the presence of the previously isolated C57BL/6 pancreatic islets (20 islets/well). RAW264 macrophages were seeded on the 5-µm pore membrane of the insert. After 24 h of incubation at 37°C and 5% CO2, all cells in the lower wells were collected, and macrophages (F4/80+ cells) were quantified using flow cytometry to measure macrophage migration.
PITx
Pancreatic islets (180 islets) isolated from C57BL/6 mice were transplanted into the livers of C57BL/6 recipient mice with or without IMCs (1.0 × 106 cells) via the portal vein 27 . In some cases, the isolated pancreatic islets were transplanted with C57BL/6 splenocytes (1.0 × 106 cells). Twelve hours after PITx, the livers of the recipient animals were collected and used for further analyses.
Histopathological Studies
Paraffin-embedded sections of liver samples from the recipient animals were deparaffinized with xylene and dehydrated with ethanol. The sections were then stained with hematoxylin and eosin or prepared for immunohistochemistry. Non-specific staining was blocked by incubation in a blocking buffer for 30 min at room temperature. Slides were then incubated overnight at 4°C with the following primary antibodies: insulin (A0564; Dako, Glostrup, Denmark) and F4/80 (MCA497R; Bio Rad Laboratories, Hercules, CA, USA). After washing, the sections were incubated with the biotinylated secondary antibodies anti-guinea pig IgG (BA-7000; Vector Laboratories, Newark, CA, USA) and anti-rat IgG (BA-9400; Vector Laboratories), respectively, for 45 min at room temperature. Visualization was performed using 3,3ʹdiaminobenzidine substrate (K3468; Dako).
Statistical Analysis
Statistical analyses were performed using the GraphPad Prism software version 9 (GraphPad Software Inc., San Diego, CA, USA). The results were presented as mean values ± standard deviation (SD). Student’s t-test was used to compare the means between two groups, and one-way analysis of variance was used to compare multiple groups. A P value of <0.05 was considered statistically significant.
Results
Cell Number and Phenotypes of Murine IMCs
IMCs were induced based on a previously described method 20 . To investigate whether the induced murine IMCs exhibit characteristics similar to human IMCs 29 , their phenotypes and immunosuppressive effects were evaluated. The cell number decreased to approximately 20% of the initial cell count after 7 days of co-culture with anti-CD80/CD86 mAbs (Fig. 2). The phenotypes of IMCs, including regulatory T cells and macrophages, did not differ significantly before or after the induction (Table 1).

Cell number during IMCs induction. During the IMCs induction period, the cell number decreased from the initial cell count (40 × 106 cells) to 8.81 ± 2.83 × 106 cells in the IMCs group and to 8.60 ± 2.68 × 106 cells in the control group cultured without mAbs. There was no significant difference between the groups (mean ± SD, n = 8).
Phenotypes of IMCs. The phenotypes of IMCs, including regulatory T cells and macrophages, showed no significant differences before and after induction (mean ± SD, n = 8).

IMCs: immunomodulatory cells, NK: natural killer, DCs: dendritic cells, Tregs: regulatory T cells.
IMCs Showed Donor-Specific Immunosuppressive Effects in the Mixed Lymphocyte Reaction
IMCs were examined to assess their immunosuppressive capacity in the mixed lymphocyte reaction (MLR). Consistent with the findings in our previous studies20,29 using human PBMCs-derived IMCs, murine IMCs effectively suppressed the proliferation of CellTrace Violet-labeled naïve responder cells (H-2b) induced in response to the BALB/c (H-2d) donor antigen in a dose-dependent manner. There was no significant suppression of responder cell proliferation against third-party (C3H/HeJ; H-2k) antigens (Fig. 3).

Inhibition of the proliferative response in the MLR. Freshly isolated responder splenocytes (C57BL/6-Ly5.1, 1.0 × 105 cells/well) were incubated with the irradiated stimulator cells (1.0 × 105 cells/well) in the presence or absence of different concentrations of IMCs. The numbers on the y-axis represent the ratio of the generated cells to the naïve responder cells: w/o = no IMCs, 1:2 = 5 × 104/well, 1:4 = 2.5 × 104/well, 1:8 = 1.25 × 104/well, 1:16 = 6.25 × 103/well. Each bar represents the mean ± SD, normalized to the percentage of proliferation of CTV-labeled naïve responder cells against the stimulator cells on day 6. (*P < 0.05. ***P < 0.001, n = 5). MLR: mixed lymphocyte reaction, IMCs: immunomodulatory cells, CTV: CellTrace Violet.
IMCs Suppressed LPS-Induced NO Production in RAW264 Macrophages
The innate immune response leads to early islet damage and impaired PITx outcomes. Therefore, we focused on macrophages to explore the anti-inflammatory potential of IMCs. To evaluate the effects of IMCs on macrophage activation, we measured NO production, a hallmark of M1 polarized macrophages known to induce islet necrosis and apoptosis 34 . We quantified the nitrite concentration in the cell culture supernatants of RAW264 macrophages after 48 h of LPS exposure. The nitrite concentration in the culture supernatant of RAW264 cells increased from 3.99 ± 0.69 µM in unstimulated controls to 67.66 ± 14.72 µM in LPS-stimulated groups (P < 0.001), confirming effective macrophage activation. In this setting, IMC treatment markedly attenuated the nitrite concentration in the cell culture supernatant of RAW264 macrophages to 47.01 ± 13.09 µM (P < 0.01, Fig. 4). Notably, the addition of freshly isolated splenocytes to LPS-stimulated macrophages did not reduce NO production (58.85 ± 15.77 µM, P < 0.56).

Nitric oxide production. RAW264 macrophages (5.0 × 105 cells/well) were co-cultured with IMCs (5.0 × 105 cells/well) and stimulated with LPS (100 ng/mL). Cell culture supernatant was collected after 48 h of culture, and nitrite production was assessed by Griess assay. Unstimulated macrophages and freshly isolated splenocytes were used as controls (*P < 0.05, n = 5). IMCs: immunomodulatory cells, LPS: lipopolysaccharide.
IMCs Suppressed the Conversion to M1 Macrophages and Promoted a Shift Toward the M2 Macrophage Phenotype
The polarization of macrophages was assessed using flow cytometry to evaluate their functional profile. After 6 h of LPS stimulation, IMCs-treated macrophages exhibited a significant reduction in the expression of the M1 macrophage markers, CD80 and CD86, compared to that in the untreated groups. Conversely, the M2 macrophage markers, CD163 and CD206, were markedly upregulated following IMC treatment (Fig. 5A, B). No significant differences were observed between the LPS-stimulated macrophages and groups treated with control cells cultured without mAbs (data not shown). Cytokine protein levels in the co-culture supernatants were quantified using ELISA. IMCs-treated groups exhibited a significant increase in the secretion of IL-10, while the production of the pro-inflammatory cytokines, TNF-α and IL-6, was reduced compared with that in the untreated controls (Fig. 5C). Notably, cytokine secretion by IMCs cultured in LPS-conditioned media without RAW264 cells was negligible and similar to that by unstimulated macrophages (data not shown).

Effect of IMCs on M1/M2 polarization. (A) RAW264 macrophages were co-cultured with IMCs (1 × 106 cells/well) and stimulated with LPS (100 ng/mL) in direct cell–cell contact model. (B) The addition of IMCs significantly reduced the percentage of M1 macrophage markers in F4/80+ gated cells. Conversely, the expression of M2 macrophage markers was increased with IMC treatment (*P< 0.05. ***P < 0.001, n = 6). (C) Culture supernatant was collected to assess cytokine production in the direct cell–cell contact model using ELISA kits. IMC treatment significantly increased IL-10 secretion compared with that in the controls (**P < 0.005), while reducing the secretion of the pro-inflammatory cytokines, TNF-α and IL-6 (*P < 0.05). IMCs: immunomodulatory cells, LPS: lipopolysaccharide, ELISA: enzyme-linked immunosorbent assay, IL: interleukin, TNF-α: tumoral necrosis factor-α.
Direct Cell–Cell Contact Is Crucial for IMCs to Exert Their Effects on Macrophages
To elucidate the mechanism underlying the effects of IMCs on macrophage polarization, IMCs were co-cultured with LPS-stimulated RAW264 macrophages using a separate co-culture model (Fig. 6A). The addition of IMCs to the upper chamber significantly suppressed the expression of the M1 macrophage markers, CD80 and CD86, compared with that in the control group. Interestingly, unlike in the cell–cell contact model, there was no corresponding increase in the M2 macrophage markers, CD163 and CD206, after treatment (Fig. 6B). Similar to the findings in the direct cell–cell co-culture model, IMCs significantly reduced TNF-α and IL-6 secretion in the culture supernatant compared with that in the untreated controls in this co-culture model. However, no significant increase in IL-10 production was seen (Fig. 6C).

Effect of IMCs on M1/M2 polarization in a separate co-culture model. (A) RAW264 macrophages were seeded in the lower chamber of a Transwell system and stimulated with LPS (100 ng/mL). IMCs were added to the upper chamber (1 × 106 cells/well). (B) The addition of IMCs significantly suppressed the expression of the M1 macrophage markers, CD80 and CD86, compared with that in the untreated controls. There was no significant difference in the expression of the M2 macrophage markers, CD163 and CD206 (*P < 0.05, n = 6). (C) Similar to the direct contact model, TNF-α and IL-6 production was reduced compared with that in the untreated controls (*P < 0.05. **P < 0.005, n = 6). However, in contrast to the cell–cell contact model, IL-10 secretion did not increase significantly with IMC treatment (P = 0.2). IMCs: immunomodulatory cells, IL: interleukin, TNF-α: tumoral necrosis factor-α.
IMCs Exerted Islet-Protective Effects in-vitro
Based on the modulatory effects of IMCs on macrophages, we investigated the impact of IMCs-mediated inhibition of macrophages using an in vitro islet co-culture model. To focus solely on the effects of the inflammatory responses exerted by activated macrophages on islets, BALB/c pancreatic islets were used in this assay. Isolated BALB/c islets were co-cultured with LPS-stimulated RAW264 macrophages, in the presence or absence of IMCs. After 12 h of co-culture, the number of remaining islets was significantly larger in the IMCs-treated groups than in the controls (76.15 ± 17.40% in treated groups vs 33.25 ± 17.46% in untreated controls, P < 0.005, Fig. 7A).
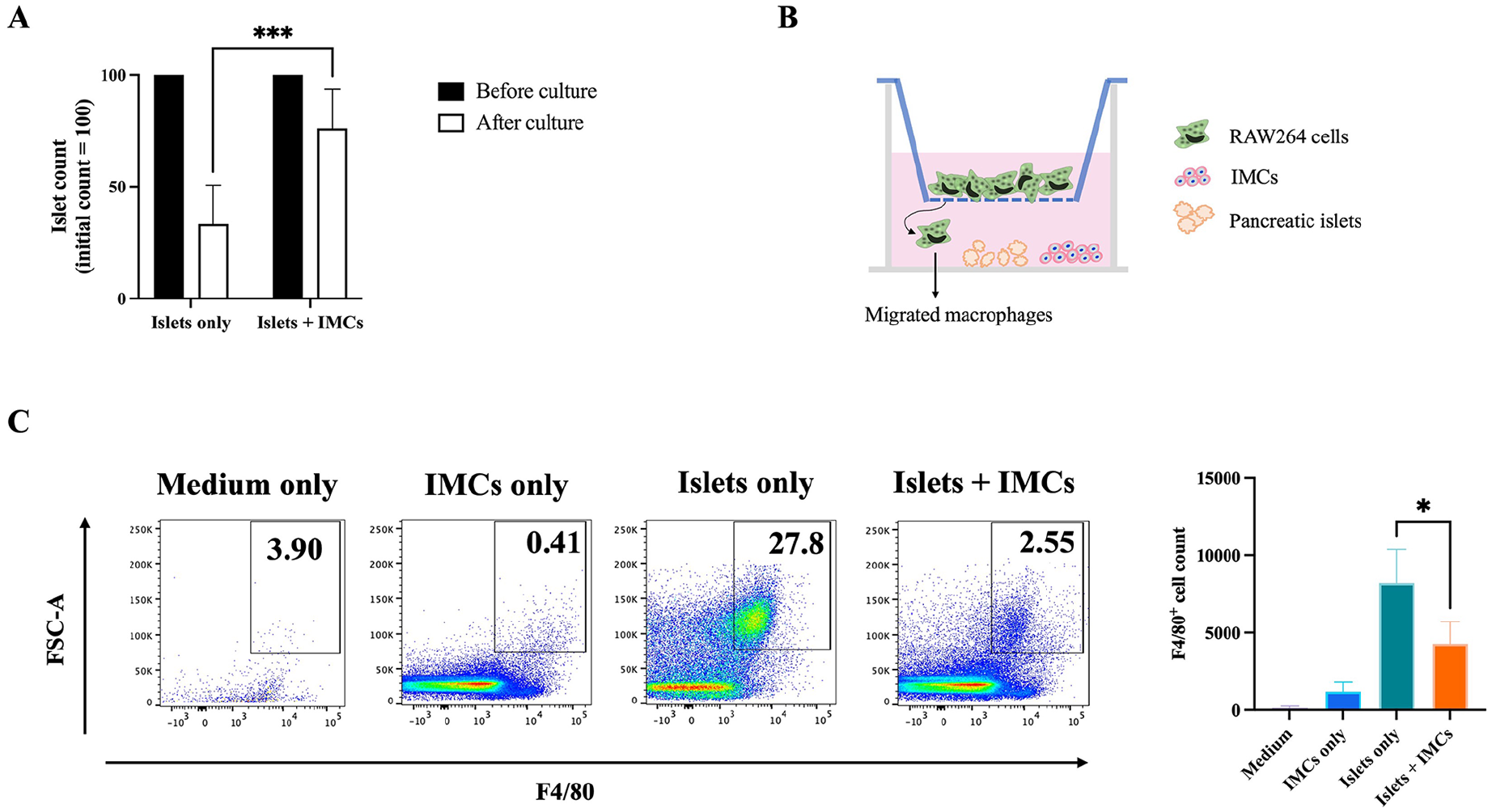
Islet-macrophage co-culture with IMCs. (A) RAW264 macrophages were stimulated with LPS (100 ng/mL) and co-cultured with IMCs (1.0 × 106 cells/well) and BALB/c pancreatic islets, and the islets were counted after 12 h of incubation. The addition of IMCs significantly prevented islet loss compared with that in the control group. The results were expressed as islet percentage considering an initial islet count of 100 (***P < 0.001, n = 4). (B and C) RAW264 cells (4.0 × 105) were seeded on top of the 5-μm porous membrane of the Transwell insert. IMCs (4.0 × 105) were seeded in the lower well with or without C57BL/6 pancreatic islets (20 islets/well). Representative flow cytometry plots showing the frequency of F4/80-positive cells present in the bottom wells in each experimental group. The addition of IMCs into the wells containing allogeneic islets significantly reduced the F4/80+ cell count in the bottom chamber (**P < 0.005). Medium: no cells or islets placed in lower wells. IMCs only: IMCs seeded in the lower well. Islets only: C57BL/6 islets in the lower well. Islets + IMCs: C57BL/6 islets and IMCs in the lower well. IMCs: immunomodulatory cells, LPS: lipopolysaccharide.
IMCs Suppressed Macrophage Migration Toward Allogeneic Islets In Vitro
To assess the effectiveness of IMCs in suppressing macrophage migration toward the islets, we employed a separate co-culture system to mimic allogeneic PITx in vitro. Given that the RAW264 cell line is derived from BALB/c mice, C57BL/6 pancreatic islets were used. RAW264 macrophages were seeded on a Transwell insert, which had a pore size sufficient to allow cell passage through the membrane, in the presence or absence of IMCs and/or C57BL/6 pancreatic islets. After 24 h of incubation, all cells in the lower wells were collected, and the number of migrated F4/80+ cells was quantified by flow cytometry. When RAW264 cells were cultured in the presence of medium only, a low number of F4/80+ macrophages was found in the bottom of the wells (126.8 ± 123.1). The presence of IMCs alone in the lower wells attracted a slightly larger number of macrophages (1187 ± 605.1), whereas the presence of C57BL/6 islets alone induced a substantial increase in macrophage migration (8212 ± 2157). In sharp contrast, when IMCs were added to the wells containing allogeneic islets, the F4/80+ cell count in the bottom chambers was significantly reduced (4285 ± 1418, P < 0.05, Fig. 7B, C).
Intraportal IMC Administration Together With Islets Reduced Inflammatory Response at the Transplantation Site After Syngeneic PITx
Finally, we evaluated whether IMC treatment reduced the inflammatory response following intrahepatic PITx. Pancreatic islets from C57BL/6 mice were transplanted into recipient C57BL/6 mice via the portal vein with or without IMCs (5.0 × 106 cells/mouse). Liver samples were collected 12 h post-transplantation, and the transplanted islets were detected inside the portal area with insulin staining (Fig. 8A). The mRNA expression of pro-inflammatory cytokines was assessed using quantitative polymerase chain reaction (qPCR). Intraportal administration of IMCs together with islets significantly reduced the mRNA expression of TNF-α and IL-1β in the liver compared with that in the controls (Fig. 8B). Immunohistochemical examination using F4/80 staining showed a significantly reduced number of F4/80+ cells in the livers of the IMC-treated groups compared with those in the controls (Fig. 8C).

Intraportal administration of IMCs in a syngeneic PITx model. Recipient C57BL/6 mice were administered 180 isolated C57BL/6 mouse pancreatic islets with or without IMCs (5.0 × 106 cells) through the portal vein. Liver samples were collected 12 h after PITx. (A) The transplanted islets were detected inside the portal area in all the transplant recipients with insulin staining. (B) IMC administration significantly reduced the mRNA expression of TNF-α and IL-1β in the liver compared with that in the control groups. Results were normalized to GAPDH housekeeping gene expression (*P < 0.05. **P < 0.005). (C) IMC administration significantly reduced the number of F4/80+ cells in the livers of the treated mice compared with that in the controls (F4/80+ cells were counted in six randomly selected sections per mouse, *P < 0.05. **P < 0.005). IMCs: immunomodulatory cells, PITx: pancreatic islet transplantation, IL: interleukin, TNF-α: tumoral necrosis factor-α, GAPDH: glyceraldehyde 3-phosphate dehydrogenase.
Discussion
In this study, murine IMCs were induced using anti-CD80 and anti-CD86 mAbs based on our previous clinical trial 20 . Similar to human PBMCs-derived IMCs used in the clinical study, murine IMCs suppressed the proliferation of naïve responder cells against donor antigens in a dose-dependent manner, thereby confirming their donor-specific immunomodulatory effects (Fig. 3). Similar to the case of solid organ transplantation, achieving immunological tolerance remains the ultimate goal of PITx. However, it is important to consider the distinctions between the immunological aspects of organ transplantation and PITx. Increasing knowledge on the inflammatory pathways involved in PITx highlights how innate immunity drives adaptive immune responses, ultimately leading to allogeneic rejection 27 . Therefore, we explored the anti-inflammatory properties of ex vivo–generated donor antigen–specific IMCs, focusing on macrophage modulation, and evaluated the potential of this novel cell therapy in the context of PITx.
When isolated pancreatic islets are infused into the recipient liver via the portal vein, the exposure to tissue factor on the islet surface triggers the release of pro-inflammatory cytokines by endothelial cells, resulting in the recruitment of leukocytes, particularly macrophages23,35. Macrophage activation is widely recognized as a key factor involved in initiating the innate immune response, leading to early islet graft loss after PITx22,25,28,36. Activated liver-resident macrophages (Kupffer cells) and monocyte-derived inflammatory macrophages secrete pro-inflammatory mediators, leading to islet damage. Kaufman et al. 37 demonstrated that the primary non-function of islet allografts is due to a cell-mediated host immune response dependent on macrophages or macrophage byproducts. Indeed, we have shown that NF-κB inhibition and an erythropoietin analogue protect islets through their inhibitory effects on macrophages, thereby improving islet engraftment and prolonging islet graft survival27,28,38. Considering the distinct challenges associated with PITx compared with solid organ transplantation 39 , it is essential to assess whether IMCs can modulate macrophage function and thereby enhance pancreatic islet survival.
We explored the anti-inflammatory potential of IMCs by focusing on their effects on macrophage polarization. We demonstrated that IMCs effectively suppressed the shift toward the pro-inflammatory M1 phenotype, while promoting a shift toward the anti-inflammatory M2 phenotype in LPS-stimulated RAW264 cells (Figs. 5 and 6). Macrophages are typically classified into two distinct polarization profiles based on the Th1/Th2 paradigm 40 . M1 macrophages, or “classically activated” macrophages, are induced by microbial products or by IFN-γ and are characterized by the secretion of pro-inflammatory cytokines, such as IL-1β, IL-6, TNF-α, and IL-12. They also express inducible nitric oxide synthase (iNOS) and the surface markers CD80 and CD86. In contrast, M2, or “alternatively activated” macrophages, are driven by IL-4 and IL-1341,42. M2 macrophages exhibit anti-inflammatory and tissue-remodeling properties by producing IL-10 and TGF-β and express the enzyme, arginase-1, along with the surface markers, CD163 and CD206. In the context of transplantation, the predominance of M2 macrophages has been observed in functioning kidney grafts, whereas M1 macrophages are more prevalent in patients with graft rejection 43 . Furthermore, the adoptive transfer of M2 macrophages has been shown to prolong islet graft survival after PITx 44 . In this study, the modulatory effects of IMCs on shifting the M1/M2 macrophage population toward M2 dominance were also correlated with an increase in IL-10 levels and a reduction in TNF-α and IL-6 production. Interestingly, co-culturing with IMCs in a separate culture setting did not enhance M2 marker expression or IL-10 secretion (Fig. 6B, C). Given that the IMCs culture supernatant on day 7 contained low levels of IL-10 (data not shown), IMCs may exert their protective effects, in part, by enhancing IL-10 secretion by M2 macrophages.
The predominant M1 or M2 macrophage population further reinforces the predominance of the corresponding macrophage phenotype within the tissue microenvironment. To assess how the IMCs-mediated modulation of M1/M2 macrophage polarization influences the functional profile of macrophages, we focused on NO, a hallmark of M1 macrophages and critical effector molecule that induces islet necrosis and apoptosis25,34. We investigated nitrite production in LPS-activated RAW264 cells cultured in the presence or absence of IMCs using the Griess assay. IMC treatment significantly reduced nitrite production by activated macrophages (Fig. 4). Kroncke et al.
25
previously demonstrated that the depletion of
There are some limitations to this study. The RAW264 cell line was employed to evaluate the effects of IMCs on macrophage activation and the inflammatory response. However, immortalized macrophages derived from tumor cells can lose macrophage-specific genotypic and phenotypic characteristics during extended culture and passaging, leading to differences compared with primary macrophages or cells in vivo. Consequently, validating the results using primary isolated macrophages would be desirable. In addition, we opted for a 12-h sacrifice time point in our PITx studies, which limited the thorough assessment of the early post-transplant islet injury. Moreover, naïve recipient mice were used in this study to avoid streptozotocin-induced liver inflammation, allowing exclusive focus on the innate immune response after PITx. Therefore, therapeutic markers such as normoglycemic rate and blood glucose levels could not be evaluated. While our findings indicate that cell–cell contact is critical in inducing the shift toward the M2 anti-inflammatory phenotype, further research is needed to identify the specific transcription factors and signaling pathways modulated by IMC treatment. Additional in vivo studies are necessary to evaluate the effect of IMCs on the inflammatory response following allogeneic PITx and to determine their potential to improve post-transplant outcomes.
Taken together, the findings in this study indicate that IMCs promote a shift from the M1 to the M2 dominant phenotype via direct cell–cell contact. Subsequently, IMCs reduce the secretion of pro-inflammatory cytokines and NO production while enhancing IL-10 secretion by M2 macrophages, ultimately favoring pancreatic islet survival. Moreover, intraportal infusion of IMCs along with pancreatic islets suppressed the mRNA expression of pro-inflammatory cytokines and abrogated macrophage infiltration in the recipient liver. Considering that the initial non-specific immune reaction is strongly associated with the subsequent adaptive immune response that leads to allograft rejection, mitigating inflammation following PITx is crucial for improving long-term graft function and survival27,55. Further in vivo studies of allogeneic PITx are necessary to gain a deeper understanding of IMC therapy before its application. These studies are currently under progress.
IMCs exert anti-inflammatory effects, particularly on macrophages. Given the combination of these effects and its intrinsic donor antigen-specific immunosuppressive capacity, IMC treatment represents a promising therapeutic approach for improving PITx outcomes.
Footnotes
Acknowledgements
The authors thank Dr Moto Fukai, Ms Nozomi Kobayashi, and Ms Awan Ajeenah for their contributions to the experiments, and Dr Makiko Kumagai-Brae for her valuable insights and suggestions. We also thank Riken BioResource Center for providing the RAW264 cell line, and Juntendo University for kindly providing the anti-mouse anti-CD80 (RM80) and anti-CD86 (GL-1) monoclonal antibodies.
Ethical Approval
All animals were cared for in strict accordance with the Guidelines for the Care and Use of Laboratory Animals of Hokkaido University Graduate School of Medicine, and the experimental design was approved by the Institutional Animal Care Committee (Approval no. 22-0048) in April 2022. This study does not contain any studies with human participants.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Japan Society for the Promotion of Science KAKENHI (grant number 17K10501).