
Abstract
In the development of cell therapy products, immunocompromised animal models closer in size to humans are valuable for enhancing the translatability of in vivo findings to clinical trials. In the present study, we generated immunocompromised type 1 diabetic Göttingen mini-pig models and demonstrated the engraftment of human-induced pluripotent stem cell–derived pancreatic islet cells (iPICs). We induced hyperglycemia with a concomitant reduction in endogenous C-peptide levels in pigs that underwent thymectomy and splenectomy. After estimating the effective in vivo dose of immunosuppressants (ISs) via in vitro testing, we conducted exploratory implantation of iPICs using various implantation methods under IS treatments in one pig. Five weeks after implantation, histological analysis of the implanted iPICs embedded in fibrin gel revealed numerous islet-like structures with insulin-positive cells. Moreover, the area of the insulin-positive cells in the pre-peritoneally implanted grafts was greater than in the subcutaneously implanted grafts. Immunohistochemical analyses further revealed that these iPIC grafts contained cells positive for glucagon, somatostatin, and pancreatic polypeptides, similar to naturally occurring islets. The engraftment of iPICs was successfully reproduced. These data support the observation that the iPICs engrafted well, particularly in the pre-peritoneal space of the newly generated immunocompromised diabetic mini-pigs, forming islet-like endocrine clusters. Future evaluation of human cells in this immunocompromised pig model could accelerate and development of cell therapy products.
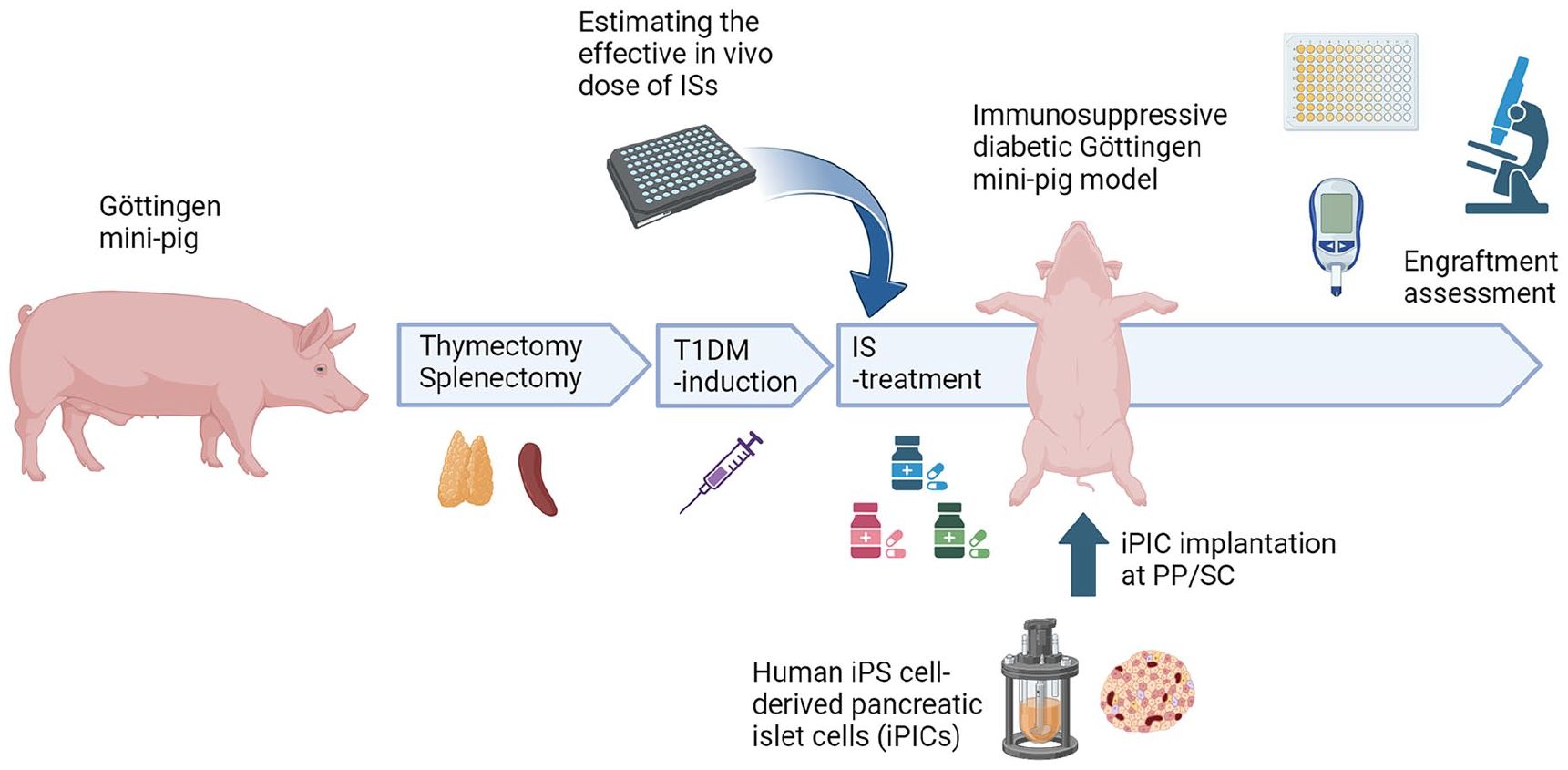
Introduction
Type 1 diabetes mellitus (T1DM) results from the destruction of insulin-releasing β-cells in the pancreatic islets. Approximately 3.3% to 13.5% of patients with T1DM experience unstable blood glucose regulation and recurrent hypoglycemic symptoms despite appropriate insulin therapy1,2. For these patients, pancreas/islet transplantation is a promising therapeutic option; however, it is hampered by the shortage and non-uniform quality of cells in clinical settings 3 . In this context, stem cell–derived islets have gained attention as an alternative cell source owing to the feasibility of large-scale cell manufacturing with uniform quality4,5.
T1DM rodent models are often used to assess the engraftment and functioning of human primary and stem cell–derived islets. However, humans and rodents exhibit significant differences in terms of anatomy, body size, and blood volume. These variations may lead to translational challenges; thus, the use of animal models closer in size to humans is preferable to obtain translatable results for clinical applications. Among the many experimental animals, dogs, non-human primates (NHPs), and pigs6,7 are commonly used. In addition, mini-pig skin resembles human skin in multiple aspects such as cell components and the thickness of individual skin layers8,9. In fact, pigs have been used to evaluate subcutaneously implanted human-sized surgical devices 10 , as well as insulin pumps, whose catheters are inserted into the subcutaneous space 11 . Therefore, diabetic mini-pigs may also be useful models for evaluating human stem cell–derived islets for diabetes treatment. However, reports on the use of diabetic mini-pigs for xenotransplantation are limited because of the complexity of xenoimmunity control in these models 12 . Itoh et al. 12 recently demonstrated the long-term accommodation of artificial human vascular grafts in mini-pigs by combining thymectomy, splenectomy, and ISs. Their findings indicate that immune-compromised mini-pigs could provide a promising platform for xenogeneic engraftment and serve as a model for evaluating the potential of cell therapy using human stem cell–derived islets, including surgical techniques in skin tissue.
Nevertheless, since both immunodeficient and diabetic states are known to render pigs severely vulnerable13,14, the feasibility of the triple combination of operational immunodeficiency, ISs, and diabetes induction is worth evaluating. Thus, the current study sought to assess the feasibility of an immunosuppressed T1DM mini-pig model and examine human islet cell engraftment in these pigs. To this end, we combined diabetes induction, immunosuppressive surgery, and the use of a triple-IS drug combination, cyclosporine (CsA), mycophenolate mofetil (MMF), and prednisolone, in Göttingen mini-pigs. After identifying the target plasma and blood concentrations of CsA and mycophenolic acid (MPA)—the active metabolite of MMF—in the pigs, we evaluated human-induced pluripotent stem cell (iPSC)-derived pancreatic islet cell (iPIC) engraftment through multiple combinations of exploratory implants and sites.
Materials and Methods
Animals
Twenty-four to 27-week-old female Göttingen mini-pigs were purchased from Oriental Yeast Co., Ltd. (Tokyo, Japan) and used for experiments (weight, ≥13 kg). The pigs were housed singly in pens under controlled conditions (temperature, 20°C–26°C; relative air humidity, 40%–70%; four air changes per hour) with a 12-h light/dark cycle. They were fed 300 to 400 g of MP-A (Oriental Yeast Co., Ltd.) daily and allowed free access to water. The experimental outline for the immunosuppressive diabetic mini-pig model is summarized in Fig. 1.
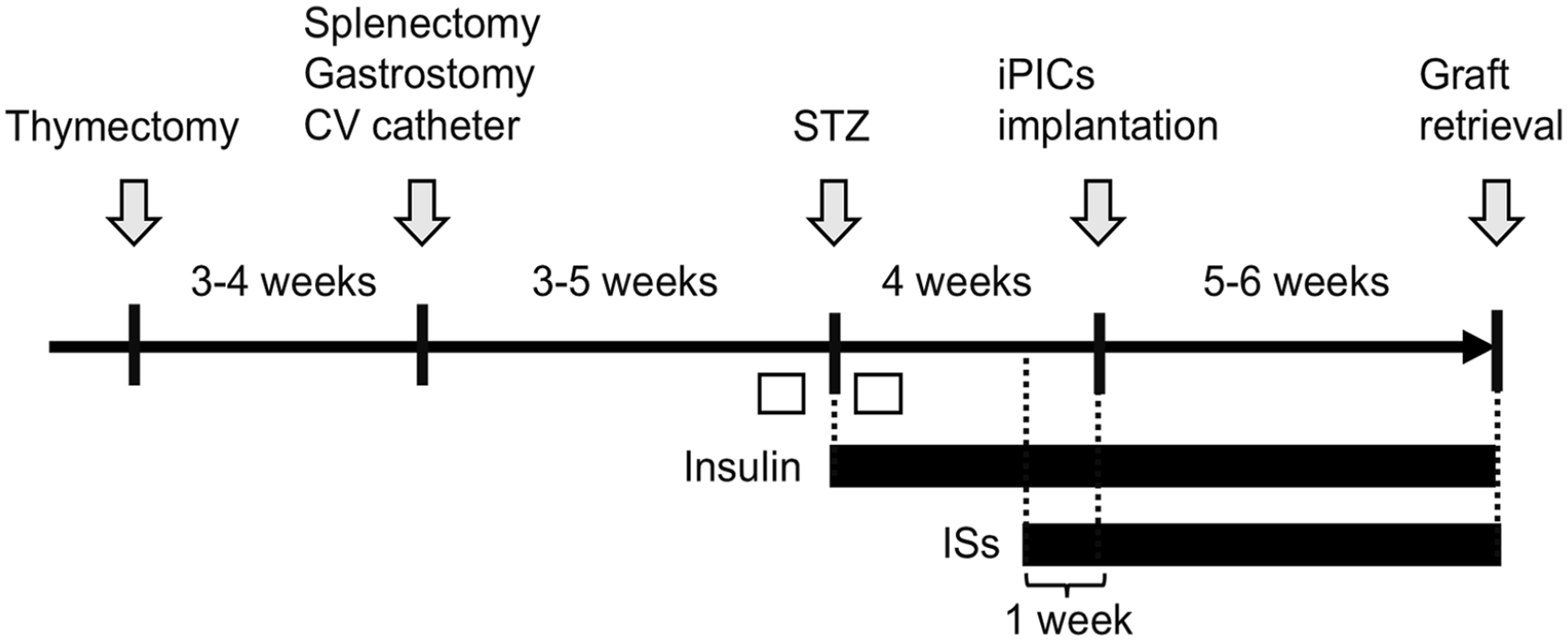
Schematic representation of the intervention in pigs. To investigate iPIC engraftment within an immunosuppressive diabetic mini-pig model, ID 1 to 3 pigs underwent the following procedures. Initially, the pigs underwent a thymectomy, followed 3 to 4 weeks later by a splenectomy and gastrostomy. A central venous (CV) catheter was placed either simultaneously or 4 weeks later. Three to 4 weeks later, streptozotocin (STZ) was administered to induce diabetes. Post-diabetes induction, continuous insulin administration was initiated. The iPICs were implanted 4 weeks after the STZ treatment. Immunosuppressants (ISs) were administered 1 week prior to the transplantation and continued throughout the experiment. An intravenous glucose tolerance test (ivGTT) was conducted before and after STZ administration (white boxes).
Establishment of the Immunosuppressive Göttingen Mini-Pig Model
The immunosuppressive Göttingen mini-pig model was established as described previously, with slight modifications 15 . Briefly, the thymus and spleen were surgically excised at 29 to 44 weeks of age. Gastrostomy was performed to create a route for delivering ISs and nutrients. A CV catheter was inserted to collect blood for pharmacokinetic studies. A 270-mg Amoxiclear tablet 100 (Kyoritsu Seiyaku, Tokyo, Japan) was administered daily to the animals after the operation. CsA (NEORAL, Nichi-Iko Pharmaceutical Co., Ltd., Toyama, Japan) 70 to 80 mg/kg/day, MMF (CellCept, Chugai Pharmaceutical Co., Ltd., Tokyo, Japan) 100 to 250 mg/kg/day, and prednisolone tablets (Teva Takeda Pharma Ltd, Tokyo, Japan) 0.16 to 1 mg/kg/day were administered through the gastric tube. The administration of ISs began 1 week before implantation and was performed while monitoring the pharmacokinetic profile and physical condition of the pigs. Hematological parameters were measured using the ADVIA 120 Hematology Analyzer (Siemens, München, Germany).
Induction of Type 1 Diabetes by STZ and Insulin Supplementation
Diabetes was induced using a single intravenous injection (200 mg/kg) of STZ (Zanosar; Nobelpharma Co., Ltd., Tokyo, Japan) through the CV catheter. Hypoglycemia resulting from acute β-cell destruction with a significant release of porcine C-peptide into the bloodstream was observed. Specifically, 20% to 30% glucose (Otsuka Co. Ltd., Tokyo, Japan) was administered via the CV catheter, or Ensure (Abbott, Chicago, IL, USA) was administered through the gastric tube when the pig blood glucose levels fell below 55 mg/dl up to 30 h after STZ injection. Blood glucose levels were monitored daily with a glucometer Accu-Chek (Roche DC, Tokyo, Japan). An ivGTT was performed before and after STZ injection. After overnight fasting, blood samples were taken at 0, 15, 30, 60, 90, 120, and 150 min following 0.5 g/kg glucose administration via the CV catheter. Blood glucose and plasma porcine C-peptide levels were measured using a glucometer and a porcine C-peptide enzyme-linked immunosorbent assay kit (Mercodia, Uppsala, Sweden), respectively. Body weight was measured twice weekly. Based on daily blood glucose levels and body weight, the long-acting insulin analogue glargine (Insulin Glargine BS Injection Kit; Fujifilm, Tokyo, Japan) was administered at 3.75 to 22.5 U/day. The area under the curve (AUC) values for blood glucose and porcine C-peptide levels in the ivGTTs were assessed before and after STZ administration.
Peripheral Blood Mononuclear Cells Proliferation Assay
Peripheral blood mononuclear cells (PBMCs) were isolated from female Göttingen mini-pigs. After 1:1 dilution of blood with phosphate-buffered saline (PBS), PBMCs were segregated by Ficoll-Paque PLUS centrifugation. The PBMCs (5 × 104 cells/100 µL/well) were seeded into a 96-well cell culture plate with RPMI-1640 containing 10% (v/v) fetal calf serum and 1% (v/v) penicillin-streptomycin. MPA or CsA was added at concentrations ranging from 0.001 to 10 µM. After pre-incubation for 30 min at 37°C, the cells were incubated with 3 µg/ml phytohemagglutinin (PHA) for 72 h at 37°C. After incubation, the plate and its contents were equilibrated at room temperature for 30 min. The culture medium was removed from each well, and Cell Titer-Glo 2.0 reagent (Promega Corporation, Madison, WI, USA) was added. The solution was then mixed on an orbital shaker to induce cell lysis. The plate was incubated for 10 min at room temperature for stabilization, and the luminescence signal was recorded using a plate reader (Perkin Elmer, Waltham, MA, USA).
Determination of Plasma Concentrations and Unbound Fractions of ISs
Blank mini-pig blood was obtained from control male mini-pigs, and blank plasma was prepared from the blood by centrifugation. The unbound fractions of the compounds in the plasma and assay medium of the PBMC proliferation assay were determined using the equilibrium dialysis method. Matrix samples were prepared by dissolving compounds in either plasma or PBMC assay medium to achieve a final concentration of 1 μmol/l.
The matrix sample was added to one side of the chamber of the HTDialysis system (HTDialysis, Gales Ferry, CT, USA) and separated by cellulose membranes (molecular weight cut-off, 6–8 kDa). An equivalent volume of PBS (pH 7.4) was then added to the other side of the chamber. The HTDialysis system was incubated at 37°C for 16 to 20 h with 8% CO2. Aliquots from both sides of the samples were deproteinized, and the concentrations of the compounds in the samples were measured using liquid chromatography-tandem mass spectrometry (LC/MS/MS). The unbound fraction was calculated as the ratio of the peak areas of the compounds in the buffer sample to those in the matrix sample.
To determine the unbound fraction of CsA in mini-pig plasma, 20% diluted plasma with saline was used for equilibrium dialysis because the unbound fraction value was low in the non-diluted plasma. The unbound fraction in non-diluted plasma, fu, was derived from the observed value obtained with 20% diluted plasma, fu′, and the following equation:
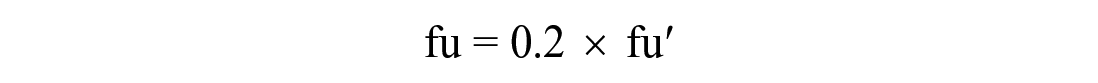
Determination of the Blood-to-Plasma Concentration Ratio
Blank mini-pig blood was obtained from male control mini-pigs. After dissolving CsA and MPA to a final concentration of 1 μmol/l, the blood was incubated at 37°C for 1 h. After deproteinization of the blood aliquot, the CsA and MPA concentrations in the blood were determined using LC/MS/MS. After incubation, the residual blood was centrifuged, and plasma was obtained. An aliquot of the plasma was deproteinized, and the CsA and MPA concentrations in the plasma were determined using LC/MS/MS. The blood-to-plasma concentration ratio of CsA was calculated as the ratio of the blood concentration to the plasma concentration.
Generation of iPICs
The human iPSC line QHJ-I14s04 was obtained from the Center for iPS Cell Research and Application, Kyoto University (Kyoto, Japan). QHJ-I14s04, an HLA-homozygous iPSC line, was established as previously described 16 . Human iPSCs were maintained on a culture surface coated with iMatrix511 (Nippi, Tokyo, Japan) using Stem Fit AK03N (Ajinomoto, Tokyo, Japan) at 37°C, 95% humidity, and 5% CO2. The cells were passaged by treatment with ethylenediaminetetraacetic acid (0.5 mM; Thermo Fisher Scientific, Waltham, MA, USA) twice weekly.
Human iPSCs were differentiated into iPICs using a previously reported method 17 with some modifications. Dissociated undifferentiated iPSCs were resuspended at a density of 2.0 × 105 cells/ml using AK03N containing 10 μM Y-27632 (FUJIFILM Wako, Osaka, Japan). The cells were cultured for 24 h to induce aggregation and were then transferred to a vertical motion bioreactor (Satake Multimix, Saitama, Japan) and cultured for 23 to 24 days. The culture medium was changed according to the following schedule.
Stage 1 (3 days)
Dulbecco’s Modified Eagle Medium (DMEM; #10569010; Thermo Fisher) containing 1× B27 supplement (with or without gamma-ray irradiation; Thermo Fisher), 1% dimethyl sulfoxide (DMSO; FUJIFILM Wako), 0.1% Pluronic F68 (Sigma-Aldrich, Saint Louis, MO, USA), activin A (10 ng/ml; PeproTech, Cranbury, NJ, USA or Ajinomoto), CHIR99021 (3 μM; Axon Medchem, Reston, VA, USA), and 1:100 penicillin-streptomycin (Thermo Fisher Scientific). CHIR99021 was added to the culture medium only on the first day. 1× B27 supplement minus insulin (with or without gamma-ray irradiation; Thermo Fisher Scientific) was used instead of 1× B27 supplement on the second and third days.
Stage 2 (4 days)
MCDB131 medium (#10372019; Thermo Fisher Scientific) containing 0.5× B27, glucose (final concentration, 10 mM; FUJIFILM Wako), NaHCO3 (1.5 g/l; FUJIFILM Wako), 1:100 GlutaMAX (Thermo Fisher Scientific), ascorbic acid phosphate magnesium salt (PMS; 58 mg/l; FUJIFILM Wako), keratinocyte growth factor (KGF; 50 ng/ml; R&D Systems, Minneapolis, MN, USA or PeproTech), 0.1% Pluronic F68, and 1:100 penicillin-streptomycin.
Stage 3 (3 days)
Improved minimum essential medium (MEM; #10373017; Thermo Fisher) containing 0.5× B27, ascorbic acid PMS (58 mg/l), KGF (50 ng/ml), LDN-193189 (100 nM; MedChemExpress, Monmouth Junction, NJ, USA), 3-keto-N-(aminoethyl-aminocaproyl-dihydrocinnamoyl)cyclopamine (0.5 μM; Toronto Research Chemicals, Toronto, Canada), 4-[(E)-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propenyl]benzoic acid (TTNPB) (10 nM; Santa Cruz Biotechnology, Dallas, TX, USA), 0.1% Pluronic F68, and 1:100 penicillin-streptomycin.
Stage 4 (4 days)
Improved MEM containing 0.5× B27, ascorbic acid PMS (58 mg/l), KGF (100 ng/ml), nicotinamide (10 mM; Stemcell Technologies, Vancouver, Canada), phorbol 12,13-dibutyrate (0.5 μM; Sigma-Aldrich), activin A (5 ng/ml), 0.1% Pluronic F68, and 1:100 penicillin-streptomycin.
Stage 5 (2 days)
MCDB131 medium containing 0.5× B27, glucose (final concentration 20 mM), NaHCO3 (1.5 g/l), 1:100 GlutaMAX, SANT-1 (0.25 μM; Sigma-Aldrich), TTNPB (10 nM), activin receptor-like kinase 5 inhibitor II (ALK5i II; 10 μM; Santa Cruz), LDN-193189 (100 nM), triiodothyronine (T3; 1 μM; Sigma-Aldrich), basic fibroblast growth factor (50 ng/ml; PeproTech or Ajinomoto), XAV939 (1 μM; Sigma-Aldrich), Y-27632 (10 μM), 0.1% Pluronic F68, and 1:100 penicillin-streptomycin.
Stage 6 (7–8 days)
MCDB131 medium containing 0.5× B27, glucose (final concentration 20 mM), NaHCO3 (1.5 g/l), 1:100 GlutaMAX, ZnSO4 (10 μM; Sigma-Aldrich), heparin sodium salt (1.4 IU/ml; AY Pharmaceuticals, Tokyo, Japan), RO4929097 (1 μM; Selleck Chemicals, Houston, TX), ALK5i II (10 μM), LDN-193189 (100 nM), T3 (1 μM), PD-166866 (1 μM; Sigma-Aldrich), 0.1% Pluronic F68, and 1:100 penicillin-streptomycin. PD-166866 was added to the culture medium and incubated for 3 days.
Stage 7 (4 days)
After 7 to 8 days of culture in Stage 6, the cells were collected and treated with TrypLE for dissociation into single cells. To induce reaggregation, the dissociated cells were cultured in a large-scale microwell bag using a previously reported method with some modifications 18 . Briefly, cells were seeded into the bag and cultured for 4 days in Stage 7 medium: MCDB131 medium containing 2% fat-free bovine serum albumin (BSA; FUJIFILM Wako), glucose (final concentration, 20 mM), NaHCO3 (1.5 g/l), 1:100 GlutaMAX, 0.5× ITS-X, ALK5i II (10 μM), T3 (1 μM), ZnSO4 (10 μM), heparin sodium salt (1.4 IU/ml), N-acetyl cysteine (1 mM; Sigma-Aldrich), Trolox (10 μM; FUJIFILM Wako), R428 (2 μM; Selleck Chemicals), PD-166866 (1 μM; Sigma-Aldrich),TR05991851 (3 μM; Takeda original multi-kinase inhibitor), Y-27632 (10 μM), and 1:100 penicillin-streptomycin.
iPIC Implantation
For the ID 1 pig, iPICs were implanted using multiple methods. For implants with fibrin gels (PP, SC1-1, SC1-2, SC2-1, and SC2-2, in Table 1), iPICs were embedded in fibrin gels, as described previously 17 . Briefly, iPICs (24,000–72,000 clusters) were mixed with 10 mg/ml fibrinogen solution (Merck Millipore, Burlington, MA, USA) and 50 IU/ml thrombin solution (Sigma-Aldrich, St. Louis, MO, USA) and incubated at 37°C for 5 min to allow for gelation. For SC-M1, polypropylene meshes (5 cm × 5 cm, Bard Mesh; Becton, Dickinson, NJ, USA) and non-woven meshes (ELTAS; Mitsui Chemicals Asahi Life Materials Co., Ltd., Tokyo, Japan) were initially implanted subcutaneously in a pig. After 48 days, iPICs embedded in the fibrin gel were implanted into the pocket formed at the implantation site. For SC-M2, iPICs were embedded in a 3% w/v alginate gel (ultrapure sterile SLG100; NovaMatrix, Sandvika, Norway) cross-linked with 70 mM SrCl2 plus 25 mM HEPES with a polyester mesh (T No. 60; Nittoku, Saitama, Japan) for reinforcement. The iPIC-alginate mixture was sandwiched by track-etched polycarbonate membranes (3 cm × 3 cm, 23-µm-thick, porous film with a pore density of 5 × 104 cm−2 and a pore diameter of 30 µm, ipPORE; it4ip, Louvain-la-Neuve, Belgium). The process for SC-M3 was similar to that for SC-M2; however, the track-etched polycarbonate membranes were coated with an MPC (2-methacryloyloxyethyl phosphorylcholine) polymer (Lipidure-CM5206; NOF Corporation, Tokyo, Japan). All fabricated materials were sterilized using ethylene oxide gas. The implants, including cells, were prepared on the day of implantation and kept in stage 7 medium at 37°C until implantation. Four weeks after diabetes induction, 360,000 iPIC clusters were implanted into the pre-peritoneal and subcutaneous spaces of an immunosuppressive Göttingen mini-pig (Table 1).
Combinations of Implant Compositions and Implantation Sites.
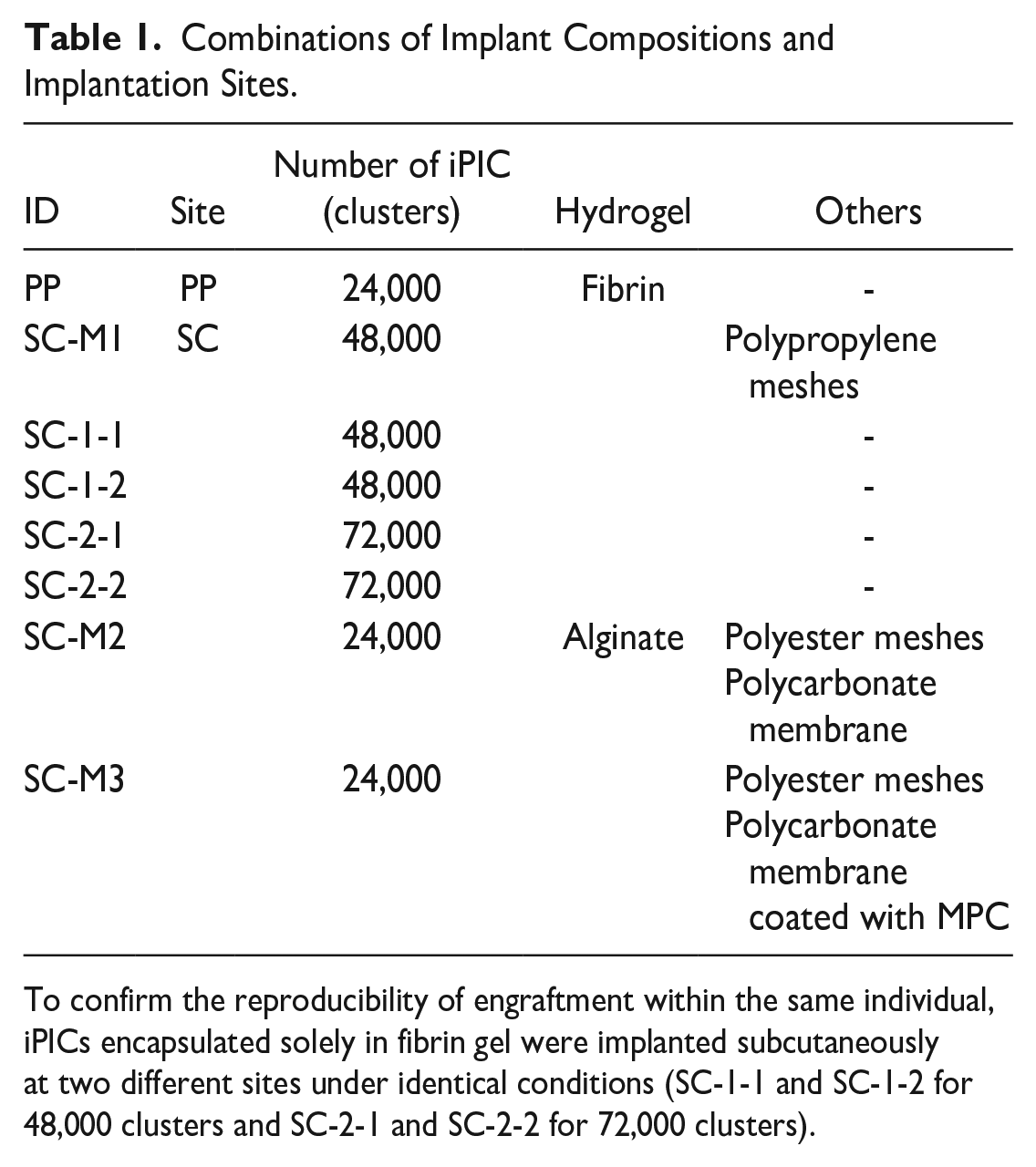
To confirm the reproducibility of engraftment within the same individual, iPICs encapsulated solely in fibrin gel were implanted subcutaneously at two different sites under identical conditions (SC-1-1 and SC-1-2 for 48,000 clusters and SC-2-1 and SC-2-2 for 72,000 clusters).
For ID 2 and 3 pigs, iPICs embedded in fibrin gels were implanted as follows: 300,000 clusters each at the PP site of a diabetic pig (ID 2) and the SC site of another diabetic pig (ID 3), respectively. A summary of the iPIC implantations is shown in Table 2.
Summary of iPICs Implantation in Diabetic Pigs.
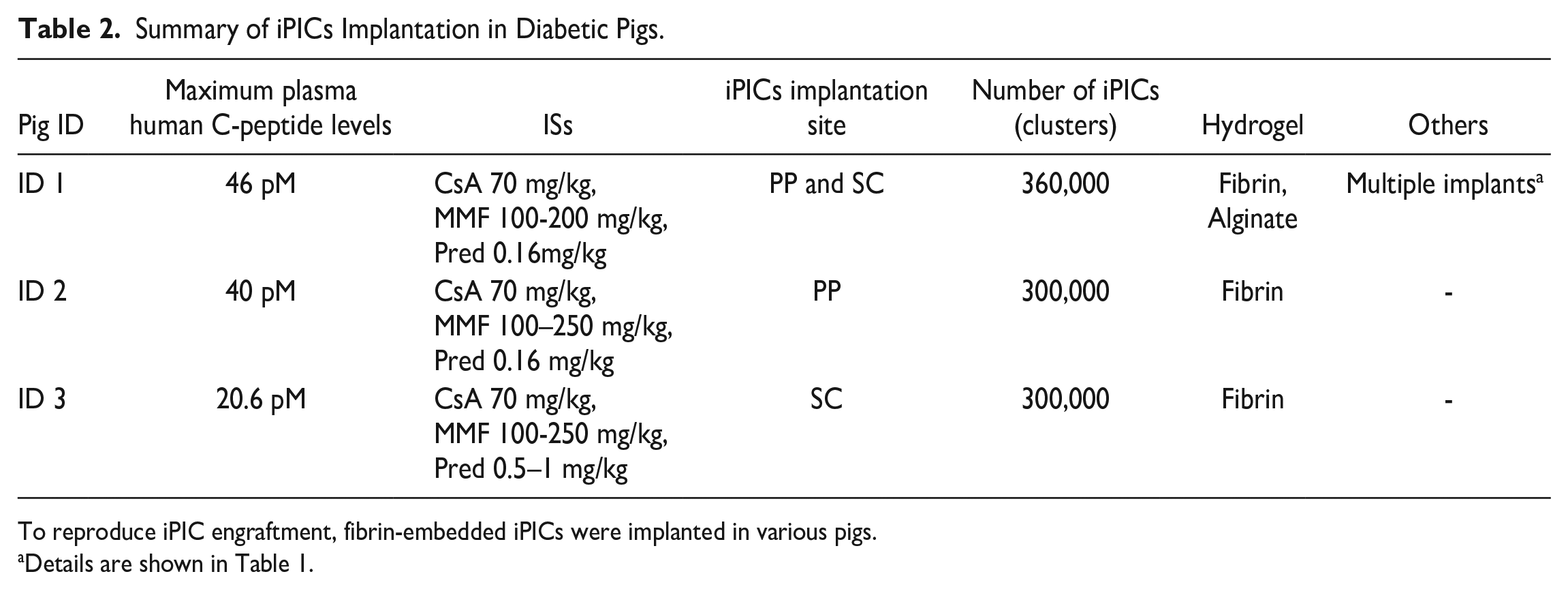
To reproduce iPIC engraftment, fibrin-embedded iPICs were implanted in various pigs.
Details are shown in Table 1.
Histological Analysis
The implanted iPIC grafts were collected 5 to 6 weeks after implantation. To identify the iPIC implantation site, a non-absorbable suture thread was used as a marker, tied around the iPIC-embedded fibrin gel. The iPICs were implanted within a 1-cm radius centered on this marker. At sites PP, SC-1-1, SC-1-2, SC-2-1, and SC-2-2, non-absorbable sutures served as skin surface markers. Tissues from both internal and external areas within a 5 cm × 5 cm zone around these suture markers were collected. All graft areas were investigated, guided by the internal marker sutures during sectioning. For SC-M1, SC-M2, and SC-M3, the non-absorbable material facilitated the identification of implantation sites by palpation. The iPICs were implanted so as to be encased by the material. The complete retrieval of the graft was confirmed during sectioning through the detection of internal markers such as sutures or materials. The retrieved grafts were fixed with 10% neutral buffered formalin for over 24 h at 4°C, and embedded in paraffin. The paraffin blocks were sliced at a thickness of 4 µm, deparaffinized with xylene three times for 5 min, and rehydrated in an ethanol gradient (100%, 90%, and 80%) three times for 5 min each. After washing with water for 5 min, the slices were stained with hematoxylin and eosin (HE) solution (FUJIFILM Wako).
For immunohistochemical analyses, the deparaffinized and rehydrated slices were soaked in epitope-retrieval buffer (Agilent, Santa Clara, CA, USA) and autoclaved at 97°C for 20 min, after which the slices were soaked in PBS for 5 min. After blocking with 0.3% Tween-20 containing 5% normal donkey serum at room temperature for 1 h, the slices were incubated with primary antibodies in Tris-buffered saline with 0.3% Tween-20 containing 5% normal donkey serum at 4°C overnight. Primary antibodies against the following proteins were used: insulin (×1/1000; GN-ID4; DSHB, Iowa City, IA, USA), glucagon (×1/200; G2654; Sigma-Aldrich Co. LLC, St. Louis, MO, USA), somatostatin (×1/200; 20067; Immunostar, Hudson, WI, USA), pancreatic polypeptide (×1/100; ab113694; Abcam, Cambridge, UK), CD3 (×1/100; ab828; Abcam, Cambridge, UK), Iba1 (×1/2000; 019-19741; FUJIFILM Wako, Osaka, Japan). The slices were then incubated with horseradish peroxidase (HRP)-conjugated or Alexa Fluor-conjugated secondary antibodies (1/500; Jackson ImmunoResearch, West Grove, PA, USA) for 1 h at room temperature; 3,3′-diaminobenzidine (DAB) was used for color development. The nuclei were stained with Hoechst 33342 (1/200, Thermo Fisher Scientific). After two additional 10-min washes in PBS, the sections were mounted using ProLong Gold antifade reagent (Thermo Fisher Scientific). For PP, SC-1-1, SC-1-2, SC-2-1, and SC-2-2, sections extending beyond 1 cm from the marker suture were processed and microscopically examined. For SC-M1, SC-M2, and SC-M3, all materials visible to the naked eye surrounding the iPICs were processed and examined under the microscope.
Human insulin–positive cells were detected automatically and quantified using HALO image analysis software ver. 3.1 (Albuquerque, NM, USA). Ten serial slides were obtained, and the number of human insulin–positive cells per classified area was counted at each implantation site. The classified area was defined by adequate coverage of the fibrous tissue and grafts thought to originate from the graft, which was determined to be 89.1 mm2. The insulin-positive cell count was normalized to the number of iPICs on the implantation day.
Statistical Analysis
A paired t-test was used to compare groups by time point, and multiplicity was adjusted using the Bonferroni method. The Welch–Aspin test was used to compare the positive area at each implantation site. P < 0.05 was considered significant.
Results
T1DM Induction After Thymectomy and Splenectomy in Göttingen Mini-Pigs
Before T1DM induction, the thymus and spleen of the mini-pigs were surgically removed to induce immunosuppressive conditions 15 . Previous reports have suggested that T1DM induction with STZ increases the risk of hypoglycemia in pig models due to massive endogenous pig insulin release accompanied by β-cell destruction in a short period 19 . Therefore, CV and gastrostomy tubes were placed not only for STZ injection or ISs treatment, but also for high-glucose administration and liquid diet to mitigate hypoglycemia. After the postoperative period, STZ was injected via the CV tube to destroy endogenous pancreatic β-cells. To identify life-threatening hypoglycemia, we monitored the changes in blood glucose and plasma porcine C-peptide levels during T1DM induction19,20. A liquid diet and glucose were administered as supplementary treatments to counteract severe hypoglycemia when blood glucose levels were lower than 50 mg/dl. The peak point of porcine C-peptide release was 15 h after STZ injection, consistent with the hypoglycemic phase in the animals tested during T1DM induction (Fig. 2A, B). In addition to controlling hyper- and hypoglycemia, because animals with diabetes mellitus are immunocompromised, the mini-pigs were treated with antibiotics and povidone iodine disinfection around the surgical incision sites every day.
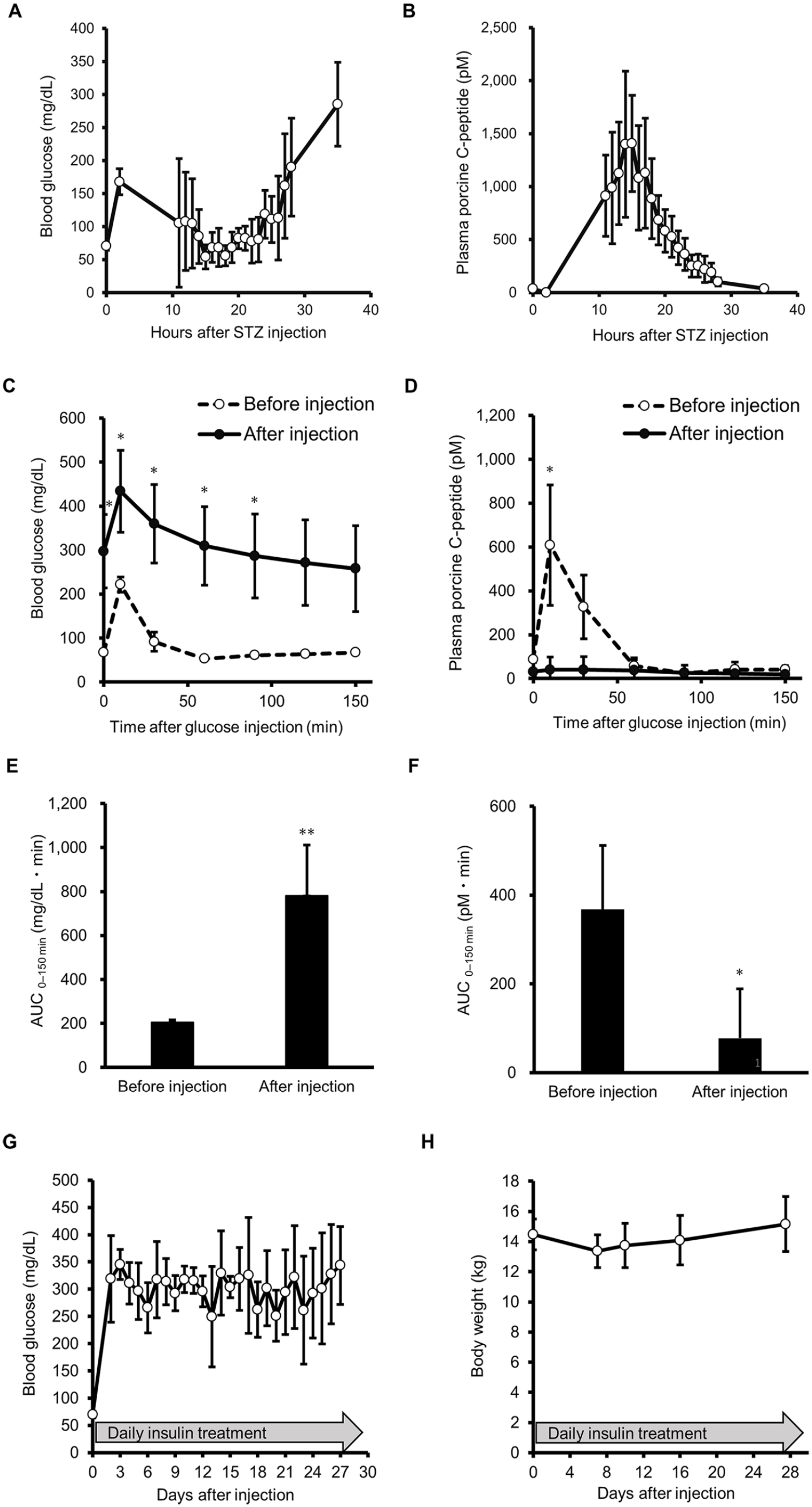
Generation of the thymectomized and splenectomized T1DM mini-pig model. (A, B) Blood glucose levels (A) and plasma porcine C-peptide levels (B) during the early period after administration of streptozotocin (200 mg/kg) to the mini-pigs. Glucose was administered as needed to avoid severe hypoglycemia during the period from 10 to 30 h after STZ injection. (C–F) ivGTTs performed before (open circle) and 3 weeks after STZ injection (closed circle). Blood glucose levels (C) and porcine plasma C-peptide levels (D) were measured after administration of glucose (0.5 mg/kg). Paired t-test was used to compare groups by time point (*P < 0.05). Multiplicity was adjusted using the Bonferroni correction method (n = 5, mean ± standard deviation [SD]). AUC for blood glucose levels (E) and porcine plasma C-peptide levels (F). Paired t-test was used to compare groups (n = 5, mean ± SD; **P < 0.01, *P < 0.05). (G, H) Fasting blood glucose levels (G) and body weight changes (H) were monitored throughout the study period.
To confirm the onset of diabetes, we evaluated glucose tolerance and plasma porcine C-peptide secretion using an ivGTT before STZ injection and 2 weeks after STZ treatment. Blood glucose levels increased ([AUC]0–2.5h: 205.1 mg·h/dl pre-STZ injection and 781.5 mg·h/dl post-STZ injection) and plasma porcine C-peptide level decreased (AUC0–2.5h: 367.7 pM·h pre-STZ injection and 77.5 pM·h post-STZ injection) after STZ treatment (Fig. 2C–F). Given that porcine C-peptide was almost completely depleted after STZ treatment, exogenous insulin was administered daily to maintain the animals’ physical condition. Fasted blood glucose levels remained >200 mg/dl at most measurement points, and body weight did not decrease throughout the observation period (Fig. 2G, H). These results suggest that diabetes was stably induced while maintaining the physical condition of the thymectomized and splenectomized mini-pigs.
Determination of the Target IS Levels in Mini-Pig Plasma and Blood
The ISs MPA and CsA are typical drugs with poor between-subject variability in pharmacokinetics 21 . To estimate the effective ranges of these drugs, we determined the target dosages of MPA and CsA in mini-pig plasma and blood. First, we examined the dose-dependent inhibition of PHA-induced proliferation of porcine PBMCs by MPA and CsA. We found that 0.3 µg/ml of MPA and 0.03 µg/ml CsA completely inhibited PHA-induced PBMC proliferation in mini-pigs (Fig. 3A, B). Next, we calculated the effective plasma concentrations. MPA and CsA exist as protein-bound and protein-unbound forms in the assay medium and plasma. The unbound fractions of MPA and CsA in the assay medium were 0.59 and 0.12, respectively, based on equilibrium dialysis 22 ; accordingly, the target unbound concentrations of MPA and CsA in the assay medium were estimated as >0.177 μg/ml and >0.0036 μg/ml, respectively, using equation (1):

According to the unbound theory, the unbound drug concentration is related to efficacy 23 . We assumed that the in vitro effective unbound concentration was equivalent to the pharmacologically effective unbound concentration in mini-pig plasma in vivo, as demonstrated in our previous report 15 . Thus, using equilibrium dialysis, the plasma unbound fractions of MPA and CsA in the mini-pigs were 0.02 and 0.006, respectively. Therefore, the target levels of the in vivo effective total concentrations in plasma were estimated to be >8,850 ng/ml and >600 ng/ml, respectively, using equation (2):

For MPA, the plasma concentration is generally considered representative of systemic exposure in clinical settings 21 . However, because CsA is mostly distributed outside the plasma, especially in erythrocytes, whole-blood levels of CsA were measured 24 rather than the plasma levels. Therefore, we aimed to monitor the blood and plasma concentrations of CsA. The in vivo target concentration of CsA in mini-pig blood was calculated to be 930 ng/ml by considering the blood-to-plasma concentration ratio of 1.55 (equation (3)):

Schematic representations of the calculation steps are shown in Fig. 3C, D.
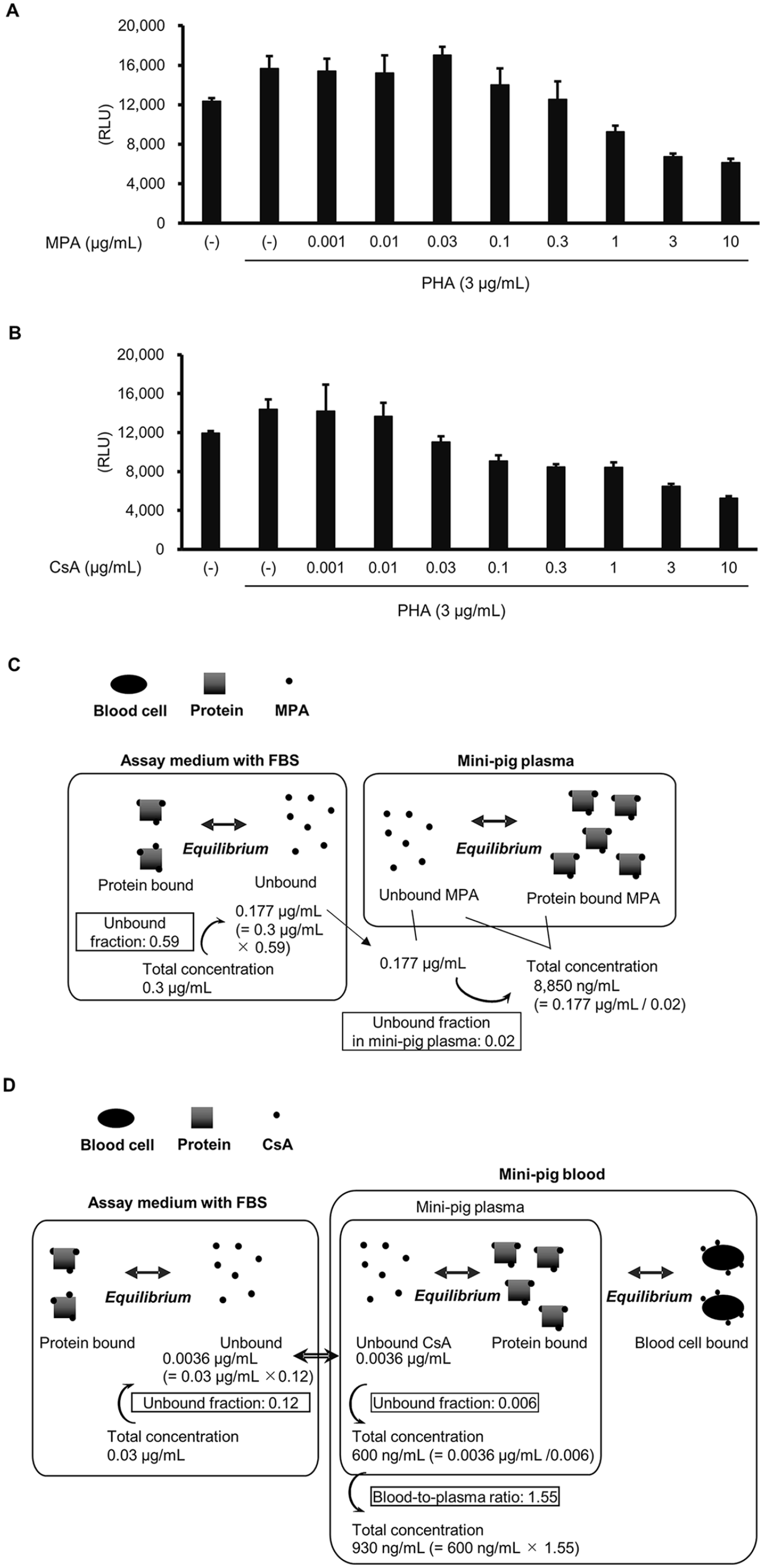
Determination of the target blood concentrations of MPA and CsA in mini-pig. MPA and CsA exist in both protein-bound and unbound forms in the blood. To determine the total effective concentrations in vivo, the unbound effective concentrations in vitro, determined using the assay media, were used. This was based on the assumption that the effective unbound concentration in vitro was equivalent to the pharmacologically effective unbound concentration in mini-pig blood in vivo. (A, B) Dose-dependent inhibition of PHA-induced PBMC proliferation by MPA (A) and CsA (B) in the assay medium in vitro. (C, D) Schematic representations of the calculation steps. The target concentration in the plasma was calculated using the in vitro-to-plasma ratio of the unbound form concentrations of MPA (C) and CsA (D). The target blood concentration of CsA was calculated using the blood-to-plasma concentration ratio. Data in (A) and (B) represent the mean ± SD of four experiments.
Engraftment of iPICs in Diabetic Mini-Pigs
During a preliminary exploration of tolerable dosages, increased doses of MMF (250 mg/kg/day) and CsA (80 mg/kg/day) caused severe side effects, such as bacterial cellulitis and thrombocytopenia. Therefore, we used 70 mg/kg/day of CsA and 100 to 200 mg/kg/day of MMF as acceptable dosages. To investigate whether our immunosuppressive treatments allowed xenoimplantation in diabetic Göttingen mini-pigs, we implanted iPICs 4 weeks after diabetes induction and 1 week after starting immunosuppressive treatments. The iPICs were implanted with hydrogels into the pre-peritoneal space, multiple subcutaneous spaces, or a single subcutaneous space with various combinations of implant compositions (Table 1). Notably, the time points at which sufficient MPA and CsA concentrations were achieved were limited during the study period (Supplementary Figure 1A and B).
To evaluate iPIC function, plasma human C-peptide levels in the fed and fasted states were monitored. Plasma human C-peptide levels were detected 7 days after iPIC implantation and gradually increased for 5 weeks, peaking at 46 pM under non-fasting conditions (Fig. 4A). No reductions were observed in the blood glucose levels and the number of daily insulin injections (data not shown).
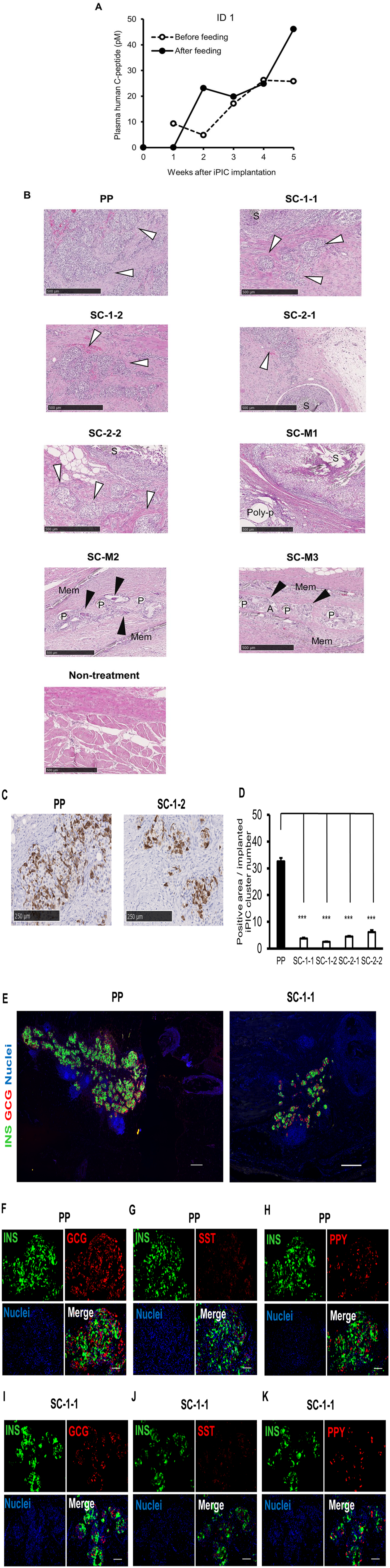
iPIC engraftment at a pre-peritoneal site and subcutaneous sites in a diabetic mini-pig. iPICs were implanted at multiple sites in the ID 1 pig using various methods. Details of these implants are described in Table 1. (A) Plasma human C-peptide levels before and after iPIC implantation. Blood samples were collected pre-feeding (open circles) and post-feeding (closed circles). (B) Representative HE-stained images of the retrieved grafts 5 weeks after iPIC implantation. “Non-treatment” indicates intact SC tissue. White arrowhead: iPICs; black arrowhead: multinucleated giant cells; S: suture; Poly-p: polypropylene meshes; P: polyester meshes; Mem: polycarbonate membrane; Alg: alginate gel. Scale bars: 500 μm. (C) Representative images of PP and SC-1-1 stained with an anti-insulin antibody and visualized with diaminobenzidine (DAB). Note: The appearance of sites SC-1-2, SC-2-1, and SC-2-2 is similar to that of SC-1-1. Scale bars: 250 μm. (D) Quantification of DAB substrate-detected human INS-positive cell counts at each implantation site, normalized to the number of clusters initially implanted. Analysis included ten sites per graft. Data are expressed as mean ± SD. The Welch–Aspin test was used for statistical comparison of positive areas at each implantation site (***P < 0.001). (E–K) Representative immunofluorescence images from the pre-peritoneal site PP (E left, F–H) and a subcutaneous site SC-1-1 (E right, I–K). Scale bars: 500 μm in E left and E right, 100 μm in F–K. INS: insulin; GCG: glucagon; SST: somatostatin; PPY: pancreatic polypeptide.
Five weeks after implantation, the grafts were collected along with the surrounding tissues to analyze the histological differences among the implantation conditions. For grafts implanted with fibrin gels without supporting materials, the number of engrafted iPICs tended to be higher in the pre-peritoneal space than in the subcutaneous spaces in HE staining and human insulin immunostaining (Fig. 4B–D, Table 3). In contrast, the other grafts showed smaller quantities or residual iPICs with massive immune cell infiltration and fibrosis (Fig. 4B). Consistently, in SC-2-1 grafts used as representative engrafted iPICs, iPICs were only slightly invaded by CD3+ T cells compared with the massive invasion in tissues around the non-absorbable suture (Supplementary Figure 3A). Meanwhile, macrophage marker iba1+ cells were moderately intermingled throughout the entire graft area, including iPICs and the suture (Supplementary Figure 3B). The insulin-positive area per cluster was higher at the pre-peritoneal site than at the subcutaneous sites (Fig. 4D), suggesting that iPICs engrafted better in the pre-peritoneal space than in the subcutaneous space. Islet-like structures were observed in the grafts at the pre-peritoneal and subcutaneous sites, primarily comprising insulin+ and glucagon+ cells, with some somatostatin+ and pancreatic polypeptide+ cells. These results indicate the feasibility of using an immunosuppressed T1DM pig model to evaluate xenoimplantation conditions (Fig. 4E–K).
Summary of HE and Immunohistochemical Pathological Analyses.
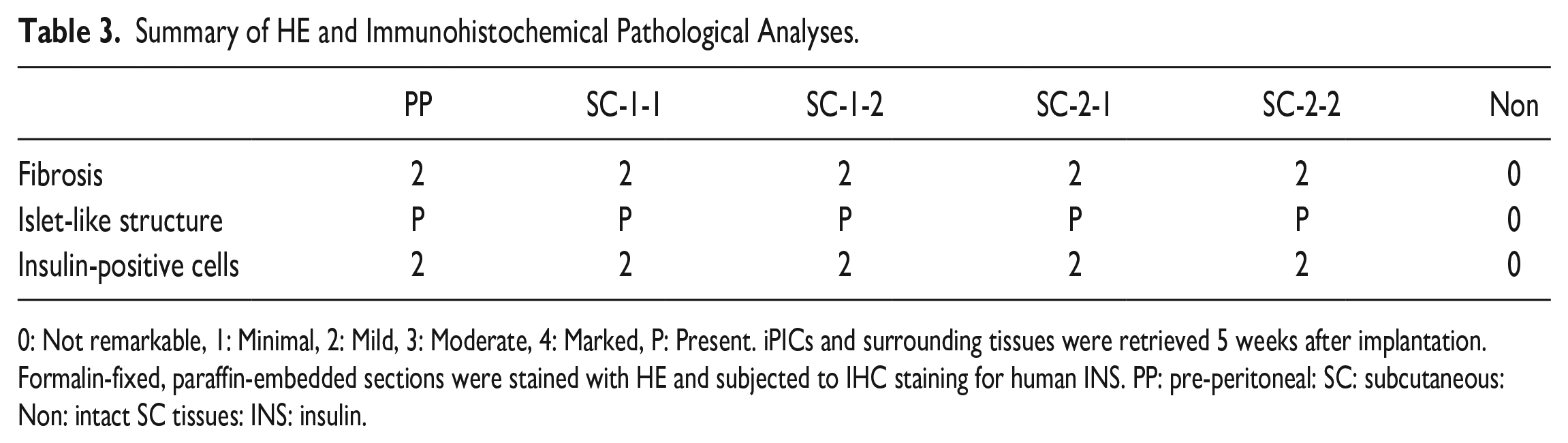
0: Not remarkable, 1: Minimal, 2: Mild, 3: Moderate, 4: Marked, P: Present. iPICs and surrounding tissues were retrieved 5 weeks after implantation. Formalin-fixed, paraffin-embedded sections were stained with HE and subjected to IHC staining for human INS. PP: pre-peritoneal: SC: subcutaneous: Non: intact SC tissues: INS: insulin.
To verify these results, we implanted the same number of iPICs into the PP site of one diabetic pig (ID 2) and the SC site of another diabetic pig (ID 3). After implantation at the PP site, plasma human C-peptide levels were detected earlier and at higher levels than the plasma human C-peptide levels detected after implantation at the SC site (Fig. 5A, B). In addition, we conducted hematological analysis on ID 2 and ID 3 to assess parameters associated with each stage of immunosuppression induction following thymectomy, splenectomy, and ISs treatment. No changes were observed in white blood cell parameters indicative of immunodeficiency, such as white blood cell count or lymphocyte counts (see Supplementary Figure 2A and B).
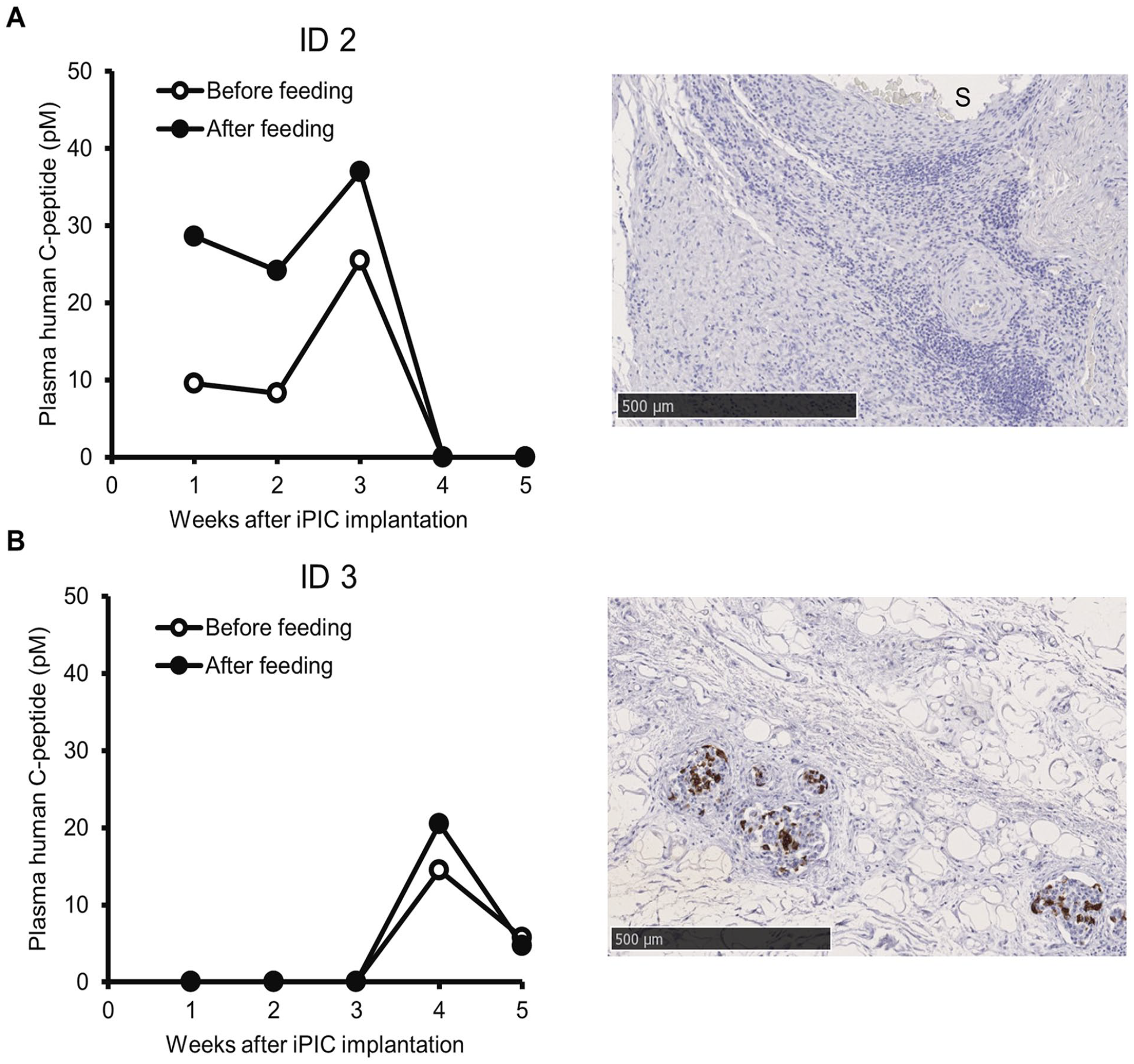
Changes in blood C-peptide levels after the implantation of a single graft into one pig. (A) iPICs were implanted into the PP site of diabetic ID 2 pig. (B) iPICs were implanted into the SC site of the diabetic ID 3 pig. Left panels indicate the plasma human C-peptide levels. Blood samples were collected before feeding (open circles) and after feeding (closed circles). Right panels show representative images of the retrieved grafts at 5 to 6 weeks after iPIC implantation. Tissue sections were stained with an anti-insulin antibody and visualized with DAB. Details of the implants are described in Table 2. Note: Plasma human C-peptide levels and images of engrafted iPICs in ID 2 pig were not obtained possibly due to an accidental infection at the implantation site approximately 1 month post-iPIC implantation.
Discussion
In this study, we implanted human iPICs into immunosuppressed diabetic mini-pigs, which represented an appropriate animal model for evaluating human-sized implants. Unlike the results obtained with small rodents, the findings in large animal models can provide useful insights related to body size, blood volume, and surgical simulation. As larger animal models, dogs, NHP, and mini-pigs have been well-studied for transplantation6,7. In particular, a mini-pig weighing 30 to 70 kg shows a smaller weight difference from humans (~60 kg) than dogs (~10 kg) and NHP (0.35–12 kg)25,26. In addition to being close to humans in body size, organ weight, and pharmacokinetics 27 , mini-pigs also show similar physical distance between tissue structures, such as the dermal layer, as humans 26 . Moreover, the amino acid sequence of the C-peptide in mini-pigs is different from that in humans, but similar to that in NHP28,29, allowing accurate estimation of insulin secretion from implanted human pancreatic islets or β-cells. Taken together, these characteristics imply that our immunocompromised diabetic mini-pigs are likely suitable for simulation and evaluation of human-sized implants, especially in terms of administration procedures and dosage.
We detected human C-peptide in mini-pig plasma 3 to 5 weeks after iPIC implantation, suggesting that the engrafted iPICs secreted insulin. However, the exact source of the secreted insulin in the ID 1 pig remained unclear, as the iPICs were implanted at multiple sites and in multiple forms to increase the possibility of engraftment. Results show that engrafted cells were rarely observed after implantation of iPICs with alginate and/or polypropylene meshes or a polyester-mesh polycarbonate membrane as components, indicating that these meshes may not have been sufficiently biocompatible after implantation in pigs. In contrast, islet-like cells were clearly observed at sites where the iPICs were embedded in fibrin gel and implanted. Thus, the cells embedded in fibrin gels and engrafted at the pre-peritoneal site and SC sites can be considered to have mainly contributed to the plasma human C-peptide detected in the mini-pig. Correspondingly, when iPICs were implanted at the PP site (ID 2) or SC site (ID 3) of the diabetic pigs, plasma human C-peptide was detected (Fig. 5A, B).
We selected pre-peritoneal and subcutaneous sites outside the peritoneal cavity as the implantation sites. The liver is the most commonly used site for islet transplantation in clinical settings because of its abundant vasculature and the need for less invasive procedures. However, islet transplantation into the liver via the portal vein has certain limitations, such as graft loss due to instant blood-mediated inflammatory reactions. Moreover, with this approach, evaluation of the transplanted islets is often difficult because of their location 30 . Therefore, multiple sites were tested to explore alternative sites and surgical procedures, including pre-peritoneal and subcutaneous sites as well as the kidney, omentum, spleen, eye, and skeletal muscle30,31. Among these, pre-peritoneal and subcutaneous sites allow implantation of higher cell doses in their large spaces and support graft retrievability, if needed, unlike the liver. Although the poor vasculature in the subcutaneous space can limit oxygen and nutrient supply, we observed some degree of cell engraftment, indicating that the iPICs used in this study could tolerate subcutaneous implantation. We speculate that this is because the islet-like cells differentiated from embryonic stem cells (ESCs)/iPSCs are relatively immature or young32,33 and require less oxygen and nutrients than adult islet cells 34 . Nevertheless, we observed more engrafted cells at the pre-peritoneal site than at the subcutaneous sites. Notably, the insulin+ cell area was more extensive at the pre-peritoneal site than that at the subcutaneous sites (Fig. 4D). In addition, when we implanted the same number of iPICs into the PP site of one diabetic pig and the SC site of another, plasma human C-peptide was detected earlier and at higher levels in the pig implanted at the PP site than the plasma human C-peptide levels detected in the pig implanted at the SC site (Fig. 5A, B). Thus, the pre-peritoneal site is likely a well-balanced implantation site for islet-like cells differentiated from ESCs/iPSCs in terms of engraftment and postoperative management.
CsA and MMF are widely used in various clinical transplantation procedures, including islet transplantation 35 (ClinicalTrial.gov identifiers: NCT00286624 and NCT00789308). Therefore, our model also offered the advantage of being able to evaluate the effects of these ISs on implanted cells. The implanted cells showed engraftment even though the in vivo MPA and CsA concentrations were lower than the target concentrations at most time points during the observation period (Supplementary Figure 1A and B). While the target concentrations represented the concentrations at which each single drug suppressed the proliferation of PBMCs isolated from mini-pigs that had not undergone thymectomy or splenectomy, the actual implantation was performed using three types of ISs along with thymectomy and splenectomy. Thus, the multiple immunosuppressive treatments may have decreased the required concentration of each drug. Clinical islet transplantation with combinations of multiple ISs has been reported previously36,37. In vitro assays with planned IS combinations would be useful for determining target concentrations that are closer to reality 15 .
This study has certain limitations. First, we were unable to conduct multiple implantation trials under identical conditions. Although the engraftment of iPICs at SC and PP sites proved reproducible in single cases, more research with large animal populations is required to conclusively determine whether the PP site is a superior implantation site for ESC/iPSC-derived islet cells. Such studies would provide the robust statistical analysis needed. Second, the implanted cells did not improve glycemic control. One possible explanation for this is that the effective number of engrafted cells was small. In addition, the observation period was insufficient to evaluate the potential of the ESC/iPSC-derived islet-like cells. In immunodeficient diabetic mice and immunosuppressed NHP, the insulin-secreting capacity increases over time after the implantation of ESC/iPSC-derived islet-like cells38,39. Thus, a higher dose or a longer observation period may have revealed greater efficacy of the implanted cells. This model may also have limited scope for evaluating vascularization and biocompatibility, since these aspects are related to the immune response and the model included immunosuppressive treatments. However, the fact that the exploratory encapsulated implants performed differently from simple fibrin gel inclusions implies that these evaluations can be performed to some extent even under immunosuppressive conditions. Indeed, pre-vascularization and biocompatibility have been assessed in immunocompromised rodents, such as SCID beige mice and nude rats 40 .
Stem cell–based therapies have also garnered considerable attention as promising treatment modalities for endocrine-related disorders beyond T1DM, including thyroid disorders, menstrual disorders, osteoporosis, and subfertility41,42. The cell implantation techniques and insights gained from this study in large animal models are applicable in pre-clinical studies to evaluate the efficacy and delivery methods of other stem cell–derived endocrine cells. Despite challenges like long-term engraftment, our findings are promising for other endocrine cell types, which benefit from high flexibility in implantation sites and minimal location restrictions.
In summary, we determined the target concentrations of MPA and CsA for immunosuppression in vitro and investigated xenogeneic implantation in the diabetic Göttingen mini-pig under combined operative and drug-induced immunosuppression. We also demonstrated the feasibility of iPIC engraftment with fibrin gel at pre-peritoneal and subcutaneous sites. With the supply of ESC/iPSC-derived islet-like cells becoming a reality, the model presented here will be useful for investigating implantation modalities in an animal system that is comparable in size to humans.
Supplemental Material
sj-tif-1-cll-10.1177_09636897241288932 – Supplemental material for Xenogenic Engraftment of Human-Induced Pluripotent Stem Cell–Derived Pancreatic Islet Cells in an Immunosuppressive Diabetic Göttingen Mini-Pig Model
Supplemental material, sj-tif-1-cll-10.1177_09636897241288932 for Xenogenic Engraftment of Human-Induced Pluripotent Stem Cell–Derived Pancreatic Islet Cells in an Immunosuppressive Diabetic Göttingen Mini-Pig Model by Midori Yamasaki, Toshiyuki Maki, Taisuke Mochida, Teruki Hamada, Saori Watanabe-Matsumoto, Shuhei Konagaya, Manami Kaneko, Ryo Ito, Hikaru Ueno and Taro Toyoda in Cell Transplantation
Supplemental Material
sj-tif-2-cll-10.1177_09636897241288932 – Supplemental material for Xenogenic Engraftment of Human-Induced Pluripotent Stem Cell–Derived Pancreatic Islet Cells in an Immunosuppressive Diabetic Göttingen Mini-Pig Model
Supplemental material, sj-tif-2-cll-10.1177_09636897241288932 for Xenogenic Engraftment of Human-Induced Pluripotent Stem Cell–Derived Pancreatic Islet Cells in an Immunosuppressive Diabetic Göttingen Mini-Pig Model by Midori Yamasaki, Toshiyuki Maki, Taisuke Mochida, Teruki Hamada, Saori Watanabe-Matsumoto, Shuhei Konagaya, Manami Kaneko, Ryo Ito, Hikaru Ueno and Taro Toyoda in Cell Transplantation
Supplemental Material
sj-tif-3-cll-10.1177_09636897241288932 – Supplemental material for Xenogenic Engraftment of Human-Induced Pluripotent Stem Cell–Derived Pancreatic Islet Cells in an Immunosuppressive Diabetic Göttingen Mini-Pig Model
Supplemental material, sj-tif-3-cll-10.1177_09636897241288932 for Xenogenic Engraftment of Human-Induced Pluripotent Stem Cell–Derived Pancreatic Islet Cells in an Immunosuppressive Diabetic Göttingen Mini-Pig Model by Midori Yamasaki, Toshiyuki Maki, Taisuke Mochida, Teruki Hamada, Saori Watanabe-Matsumoto, Shuhei Konagaya, Manami Kaneko, Ryo Ito, Hikaru Ueno and Taro Toyoda in Cell Transplantation
Footnotes
Acknowledgements
The authors sincerely thank S. Yamanaka, and K. Osafune (CiRA), as well as Y. Kajii and S. Izumo (Takeda Pharmaceutical Company), for supporting the collaborative research between Takeda Pharmaceutical and CiRA (T-CiRA). We appreciate N. Yamazoe, K. Sakuma, H. Sugiyama, A. Kuwano, S. Inoue, A. Osawa, A. Takahashi, A. Makita, M. Ohra, S. Seki, and S. Hokaiwado (Takeda Pharmaceutical Company and Orizuru Therapeutics); H. Yoshioka, T. Shimada, M. Ikehata, M. Mukaitani, and T. Kitaura (Axcelead Drug Discovery Partners); H. Hiyoshi, H Matsumoto and T. Kakizume (Takeda Pharmaceutical Company), T. Anazawa (Graduate School of Medicine of Kyoto University) for their assistance with several experiments and discussions. We would like to thank Editage (![]() ) for English language editing.
) for English language editing.
Author Contributions
M.Y., T.M., T.M., S.W-M., S.K., and T.H. performed the experiments, collected and analyzed the data, and drafted the manuscript. H.U., M.K., R.I., and T.T. provided critical advice on the research strategy, and reviewed and edited the manuscript.
Ethical Approval
The use of the iPSC line was approved by the Ethical Review Committee of the Shonan Health Innovation Park (Fujisawa, Kanagawa, Japan) and Kyoto University (approval number: #CiRA18-27).
Statement of Human and Animal Rights
Animal experiments were approved by the Institutional Animal Care and Use Committee of Shonan Health Innovation Park (Shonan iPark), Takeda Pharmaceutical Company.
Statement of Informed Consent
Written informed consent for publication in this study was obtained from homozygous HLA donors in the human iPSC stock project.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: T.T. is a scientific advisor receiving an advisory fee from Orizuru Therapeutics. The remaining authors declare no conflicts of interest.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by the T-CiRA budget from Takeda Pharmaceutical Company. This research was supported in part by AMED under Grant Number JP21be0404008 and the iPS Cell Research Fund.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.