
Abstract
Immune cell therapy as a revolutionary treatment modality, significantly transformed cancer care. It is a specialized form of immunotherapy that utilizes living immune cells as therapeutic reagents for the treatment of cancer. Unlike traditional drugs, cell therapies are considered “living drugs,” and these products are currently customized and require advanced manufacturing techniques. Although chimeric antigen receptor (CAR)-T cell therapies have received tremendous attention in the industry regarding the treatment of hematologic malignancies, their effectiveness in treating solid tumors is often restricted, leading to the emergence of alternative immune cell therapies. Tumor-infiltrating lymphocytes (TIL) cell therapy, cytokine-induced killer (CIK) cell therapy, dendritic cell (DC) vaccines, and DC/CIK cell therapy are designed to use the body’s natural defense mechanisms to target and eliminate cancer cells, and usually have fewer side effects or risks. On the other hand, cell therapies, such as chimeric antigen receptor-T (CAR-T) cell, T cell receptor (TCR)-T, chimeric antigen receptor-natural killer (CAR-NK), or CAR-macrophages (CAR-M) typically utilize either autologous stem cells, allogeneic or xenogeneic cells, or genetically modified cells, which require higher levels of manipulation and are considered high risk. These high-risk cell therapies typically hold special characteristics in tumor targeting and signal transduction, triggering new anti-tumor immune responses. Recently, significant advances have been achieved in both basic and clinical researches on anti-tumor mechanisms, cell therapy product designs, and technological innovations. With swift technological integration and a high innovation landscape, key future development directions have emerged. To meet the demands of cell therapy technological advancements in treating cancer, we comprehensively and systematically investigate the technological innovation and clinical progress of immune cell therapies in this study. Based on the therapeutic mechanisms and methodological features of immune cell therapies, we analyzed the main technical advantages and clinical transformation risks associated with these therapies. We also analyzed and forecasted the application prospects, providing references for relevant enterprises with the necessary information to make informed decisions regarding their R&D direction selection.
Introduction
Cancer is a systemic disease that exploits multiple escape mechanisms to evade anti-cancer immunity 1 and significantly change the function and composition of the immune system as a whole 2 . The evolution of anti-cancer therapies spans over a hundred years, with an ever-increasing efficacy and specificity in eliminating cancer cells while minimizing damage to healthy tissues. Toward this objective, immune cell therapy stands out among the existing approaches as a game-changing solution. In 2017, the Food and Drug Administration (FDA) approved two chimeric antigen receptor-T (CAR-T) cell therapies, axicabtagene ciloleucel (Yescarta) 3 from Kite Pharm and tisagenlecleucel (Kymriah) 4 from Novartis, for the treatment of lymphoma 5 . These approvals marked a significant milestone for the accelerating development of anti-cancer treatments.
The fundamental concept of immune cell therapy is to boost immune responses by externally supplying cells with the desired functionality to the patient for combatting cancer. It not only reinvigorates pre-existing immune responses but also drives new immune responses to the anti-tumor effort. The response of immune cell therapy commonly involves the activation and cooperation of multiple immune cell types, including antigen-presenting cells, such as dendritic cells (DCs) and macrophages, T lymphocytes (T cells), and natural killer (NK) cells. A tumor-burdened immune system is not only imbalanced in the number of immune cells but also exhibits functional abnormalities in the molecules and signaling pathways that regulate the anti-cancer immune response. Therefore, precise administration of appropriate therapeutic approaches is necessary to prevent suboptimal efficacy in immune cell therapy.
Depending on the cancer types and their mechanisms, various domains of the immune process offer avenues for therapeutic intervention. Approaches encompass, for instance, extraction and expansion of immune cells with prior cancer recognition (e.g., tumor-infiltrating immune cells) 6 , external differentiation of effective cell types [e.g., cytokine-induced killer (CIK) cells] 7 , and genetically modified cells to achieve specific recognition and tumor cell elimination [e.g., T cell receptor (TCR) engineering, chimeric antigen receptor (CAR)]8,9. In this review, we will address the state-of-the-art immune cell therapies, summarize strategies applied to enable and optimize the various categories of these therapies, and offer future perspectives.
Treat Cancer With Low-Risk Immune Cell Therapies
Low-risk immune cell therapies are designed to use a patient’s own immune cells to eliminate cancer cells. It should be noted that “low risk” emphasizes the comparatively safer and fewer adverse effects of certain therapies within the field of cell therapy, which falls under regenerative medicines or advanced therapy medicinal products (ATMPs). The category of low-risk immune cell therapies encompasses four pivotal techniques followed by: tumor-infiltrating lymphocytes (TIL) therapy, CIK cell therapy, DC vaccine, and DC/CIK cell therapy. To acquire a more comprehensive understanding of these methodologies, it is imperative to delve into the fundamental mechanisms and clinical advancements that facilitate each of these cell therapies.
TIL Cell Therapy
TILs are natural mononuclear cells found within the solid tumor microenvironment (TME), including intratumoral tumor infiltrating lymphocytes (iTILs) and stromal tumor infiltrating lymphocytes (sTILs). The sTILs are primarily effector memory T cells and are more easily detectable compared with iTILs 10 . As a group of heterogeneous lymphocytic cells that have been exposed to the tumor tissue, TILs are “pre-trained” to attack cancerous cells while leaving the healthy ones alone. In mouse tumor models, these heterogeneous cells have demonstrated potential in treating advanced cancers11,12, and the first human TIL therapy resulted in a significant regression in metastatic melanoma 13 . Solid tumors lack ideal tumor markers, making the targeting of tumor-associated antigens (TAAs) challenging14,15. TILs, being polyclonal cells with diverse receptors, provide unique treatment options for solid tumors16,17. Compared with genetically modified immune cells, TILs exhibit superiority in overcoming heterogeneity in treating solid tumors, especially highly heterogeneous tumors like melanoma18,19. In addition, they possess chemokine receptors crucial for migration toward the TME after injection, and their lower off-target toxicity compared with CAR-T cells is attributed to negative selection during T cell maturation 20 .
TILs can be isolated from resected tumor tissue. Given the sparsity of TIL in the native TME, the number of TILs available for amplification is limited, as well as additional factors that hinder the anti-tumor cytotoxicity of TIL (e.g., Treg, tumor-associated macrophages, myeloid-derived suppressor cells, and various immunosuppressive molecules21,22). The production and expansion of TILs are crucial steps for the success of TIL therapy. The expansion for TIL in vitro involves rapid expansion protocol (REP). In the pre-REP phase, TILs undergo primary expansion with high-dose interleukin (IL)-2 23 . Some methods involve selecting tumor-specific TILs for further expansion, while others expand bulk TILs to maintain efficacy. During the REP phase, both high-dose IL-2 and anti-CD3 were administered, and irradiated allogeneic peripheral blood mononuclear cells (PBMCs) were deployed as feeder cells until sufficient cells were obtained. Then the expanded cell products, passing quality controls, are ready for patient administration after lymphodepletion.
Since Rosenberg et al. 13 successfully applied TIL therapy to patients with metastatic melanoma in 1988, a series of clinical trials have emerged. In 1996, TIL therapy was first introduced to treat non-small cell lung cancers (NSCLCs) in a clinical study involving 113 patients 24 . The outcome reveals TIL in combination with IL-2 as an effective treatment for NSCLCs, with a selective benefit for stage IIIb NSCLCs compared with stage II or IIIa. Following the initial success, subsequent developments of TIL therapies have been directed toward improving efficacy in treating other tumor types, including skin 25 , renal 26 , and gastric cancers 27 . Specific approaches within these developments aim to elucidate the composition of TIL cell types and their relevance to the clinical outcome. For example, the successful generation of tumor-infiltrating B lymphocytes (TIBs) has been found to predict an improved clinical outcome in patients with metastatic renal cell cancer (mRCC) 26 . Another study of TIL treatment for gastric cancer reveals histological subgroups of TILs are associated with distinct survival time 27 . Currently, the clinical transfer of TIL therapy is still at a preliminary stage, lacking long-term clinical observations. However, the preliminary success suggests an opportunity to develop promising therapeutic TILs with more efficient isolation and expansion methods, and prospectively alternative combinations with other anti-tumor therapies.
CIK Cell Therapy
CIK cells are heterogeneous immune effector cells with a mixed T- and NK cell-like phenotype. They are generated by ex vivo incubation of human PBMC or cord blood mononuclear cells with interferon gamma (IFN-γ), anti-CD3 antibody, recombinant human IL-1 and IL-2. CIK cells uniquely exhibit major histocompatibility complex (MHC)-unrestricted targeting of tumors, reducing alloreactivity, and enabling attacks on various tumor types 3 . During the in vitro cultivation process, CIK cells secrete various cytokines to activate the cytotoxic activity of macrophages, NK cells, and CD8+ T cells, thereby directly inhibiting the growth of tumor cells and promoting indirect killing effects. CIK cells induce the expression of apoptosis genes and anti-tumor genes, conducting cytotoxic activity and facilitating tumor cell apoptosis. Their tumor-killing effects are mainly via the mechanism of binding to antigens on tumor cells through the adhesion molecule lymphocyte function-associated antigen-1/cognate ligand intercellular adhesion molecule-1 (LFA-1/cam-1). This promotes the expression of MHC-I or MHC-II molecules, leading to enhanced presentation, activation, recognition, and direct killing of tumor cells.
Activated CIK cells secrete various cytokines, including IL-2, IL-6, tumor necrosis factor α (TNF-α), granulocyte macrophage colony-stimulating factor (GM-CSF), and other cytokines, not only directly enhance cytotoxic effects and inhibit tumor cells but also indirectly kill tumor cells by regulating the immune system. IFN-γ, which is largely generated by CIK cells, enhances the activity of NK cells, macrophages, and cytotoxic T lymphocytes (CTL). It can also promote the expression of MHC-I molecules on tumor cells, leading to increased killing of CTL. Recent research 28 indicated that the secretion of IFN-γ by CIK cells could promote the expression of ICAM-1 on chronic lymphocytic leukemia (CLL) cells, positively impacting the cytotoxic effector cells’ apoptosis induction. The apoptosis genes of tumor cells can also be activated by CIK cells, as these cells express FasL, a Fas ligand that belongs to the TNF superfamily, which induces tumor cell apoptosis 29 . Verneris 28 suggested that certain tumor cells (such as melanoma and ovarian cancer) escape immune clearance by inducing lymphocyte apoptosis through FasL, but they are sensitive to CIK cells. This is because, during the induction process of CIK cells, Fas-sensitive or activated T cells are selectively eliminated by the effects of cytokines, such as IFN-γ or activation-induced cell death mechanisms. Combined with high levels of expression of anti-apoptotic genes in CIK cells, these factors allow CIK cells to tolerate apoptosis induced by tumor cells expressing FasL.
Despite concerns about recurrence and unresponsiveness, CIK cells’ heterogeneous nature allows them to target and kill various tumor cells 30 . Since the introduction of CIK cell therapy in 1991, over 5,000 patients with diverse tumor types, including lung 31 , breast 32 , brain 33 , and colon 34 cancers have participated in CIK cell therapy. The clinical trials of CIK cells (or CIK cells combined with DCs) have exceeded 80 globally35,36. The therapeutic potential of CIK cells extends beyond their interaction with NKG2D, incorporating the axis of antibody-dependent cell-mediated cytotoxicity (ADCC)37,38. Alongside other immunotherapies, conventional chemotherapies, or radiotherapies, CIK therapy suggested a synergistic anti-tumor effect 39 , evolving and is an encouraging avenue for the treatment of cancer. While the clinical landscape is promising, some open discussions are necessary. Comparisons of CIK cell toxic effects with other immune cells post-infusion, minimizing additional supportive care, determining advantages over other immunotherapies, understanding key determinants for crosstalk with CIK cells in the cancer microenvironment, and generalizing the approach across multiple centers require exploration30,40.
DC Vaccine
Manipulating a patient’s autologous DCs for “vaccination” against tumor tissues is a promising approach. DC vaccines involve treating in vitro-cultured DCs with TAAs, such as peptides, proteins, DNA/RNA, or viruses. The DCs will uptake the antigens and then active during the carefully controlled culture process. DCs serve as potent antigen-presenting cells, playing a crucial role in influencing the immune system. While immature DCs process foreign antigens, triggering maturation and migration to lymph nodes, mature DCs can foster tumor tolerance within the TME, potentially leading to T-helper type 2 responses 41 .
DC vaccines, often employing autologous monocytes matured in vitro and pulsed with antigens, have shown good tolerability in diverse tumor patients with minimal toxicity. However, the magnitude and duration of antigen-specific immune responses have generally been weak, limiting objective clinical responses. Despite limited responses in pediatric tumors, ongoing research focuses on enhancing each step of vaccine production. Strategies include expanding DC sources, improving immunogenicity, optimizing antigen selection, developing new immune adjuvants, and exploring concomitant immunomodulation or chemotherapy42,43.
Sipuleucel-T, the only FDA-approved autologous DC vaccine, offers hope for effective adult malignancy treatment, specifically for castrate-resistant prostate adenocarcinoma 44 . Manufactured by culturing a patient’s PBMCs with a recombinant protein (PAP-GM-CSF) consisting of a TAA [prostatic acid phosphatase (PAP)] and GM-CSF. This pioneering vaccine represents a significant stride in the field of DC vaccines, showcasing potential avenues for improved outcomes in both adult and pediatric malignancies 45 . In clinical trials, DCs have been used primarily in combinatory immunotherapies with CIK cells, as well as immune checkpoint inhibitor (ICI) and chemotherapy and radiotherapy to improve outcomes. Studies have shown the ability of DCs to activate natural killer T (NKT) cells 46 or to simultaneously activate CD4+ and CD8+ T cells 47 .
DC/CIK Cell Therapy
When multiple low-risk immune cell therapies are used together, a positive combined synergistic effect can be achieved, particularly evident in the co-application of DCs and CIKs adaptable and synergized. This synergism aims to enhance the body’s immune response against diseases.
CIK cells, known for their broad-spectrum tumor-killing effects 48 , face limitations due to lack of specific recognition capabilities. DCs are currently known as the most potent antigen-presenting cells, recent research indicates that the interplay between CIKs and DCs induces alterations in the expression of immunostimulatory surface molecules within both populations. Indeed, co-culturing DCs with CIK cells plays a complementary role, mutually promotes maturation, jointly increases the anti-tumor activity, and demonstrated promising synergistic effects 49 , even on tumor cell lines that were previously resistant to CIKs and were not co-cultured with DCs before 50 . The secretion of cytokines, such as IL-2, IL-12, and IFN-γ by DCs enhances the maturation of CIK cells, resulting in increased levels of CD3+, CD8+, and CD56+ subsets in CIK cells 49 . Notably, substantially increased secretion of IL-12 by the DCs significantly enhances the cytolytic function of CIKs 51 . In addition, this cooperative environment also reduces the secretion of IL-10 and the number of regulatory T cells (CD4+CD25+ Treg cells), weakening their inhibitory effect on tumor immunity and enhancing the tumor-killing effect of CIK cells. Simultaneously, CIK cells enhance the antigen presentation specificity of DCs and the expression of co-stimulatory molecules, contributing to an overall increase in anti-tumor activity. Furthermore, co-culturing of DC and CIK cells inhibits the expression of human telomerase reverse transcriptase (hTERT) protein, curbing telomerase activity and impeding tumor cell proliferation 52 .
Based on the above mechanisms, DC–CIK therapy, characterized by mutual adaptability and synergy, has become a focus of research in the field of tumor therapy 53 in recent years. Both basic and clinical researches have rapidly developed and demonstrated effectiveness in significantly enhancing anti-tumor immunity. Despite concerns about mild graft-versus-host disease (GvHD), trial experiences indicate manageable outcomes 54 . Case studies have demonstrated safety and improved efficacy55–57. DC/CIK cell therapies target various aspects of the immune system, resulting in a more comprehensive approach to fighting diseases. This is especially beneficial in complex conditions or refractory malignancies where multiple immune dysfunctions play a role.
Overall, low-risk immune cell therapies can help boost the immune system’s ability to recognize and eliminate harmful entities, resulting in improved health outcomes. The synergistic effects of these therapies have the potential to significantly improve the effectiveness of immune-based treatments and provide better outcomes for patients. However, it is crucial to evaluate and customize the precise combinations and dosages of these therapies to guarantee their safety and effectiveness for individual patients.
The Technological Progress of High-Risk Immune Cell Therapies
High-risk immune cell therapies utilize either autologous stem cells, allogeneic or xenogeneic cells, or genetically modified cells, all of which require further manipulation and evaluation. The design of these therapies is well suited to address their intended purposes. These treatments stimulate new immune responses against tumors and often have the potential to reinvigorate pre-existing ones. Here, we describe the feature design, transfection method, cell source, and methods to overcoming suppressive TME in high-risk immune cell therapies, including CAR-T, T cell receptor-T (TCR-T), CAR-NK, and CAR-macrophages (CAR-M). In addition, we offer insights into upcoming trends.
The Design of CARs
CARs are engineered receptor proteins that offer immune cells a novel ability to identify a specific antigen on the surface of cancer cells, thereby potentially triggering immune cells’ proficiency to eliminate cancer cells. Current studies suggest that even minor modifications to the CAR construct can significantly impact the therapeutic outcome 58 . It is typically composed of three modular domains: extracellular domain, transmembrane domain, and intracellular domain.
Extracellular domains: The art of targeting
The extracellular domain includes an ScFv for target binding and a hinge region, commonly immunoglobulin (Ig)-like domain hinges in CAR-engineered cells, and is anchored to the cell membrane through the transmembrane domain. Novel features that drive immune cell activation, including T cells, NK cells, and macrophages, were introduced via the extracellular domain of CARs. CAR-engineered immune cells can recognize cancer-specific target epitopes without the limitations of MHC and co-receptors, making them more effective in finding and killing cancer cells.
CAR-engineered immune cells have evolved from targeting one single antigen to targeting two different antigens for the optimization of treating negative antigen cancer cell escape or relapsed with loss antigen 59 , and binding to either antigen will trigger the activation of CAR-engineered immune cells. This can be achieved in several ways (Fig. 1A–C), such as transducing a single bicistronic vector encoding two independent CAR molecules into a single cell, or transducing a vector encoding a bivalent CAR molecule into the cells 59 , or co-administration of two different CAR-engineered immune cell products, or co-transduction of two vectors encoding different CARs into a single immune cell 60 . These approaches are particularly promising for B cell lymphoblastic leukemia, especially for the relapse or refractory B-cell acute lymphoblastic leukemia (B-ALL), as clinical studies have indicated significant efficacy of CD19/CD22 co-transduced CAR-T cells61,62 and CD19/CD22 bispecific CAR-T cells. 63 Similarly, a phase I clinical trial showed that the bispecific CD20/CD19 CAR-T could also be used to treat patients with relapsed and refractory B cell malignancies, with an overall response rate of 82% after 28 days of treatment, which showed both safe and effective for therapeutic purposes 64 . In addition, Wang et al. 65 demonstrated that CD19/CD22 bicistronic CAR-T exhibited equivalent or better function compared with monospecific CD19 or CD20 CAR-T cells in vitro and in vivo.
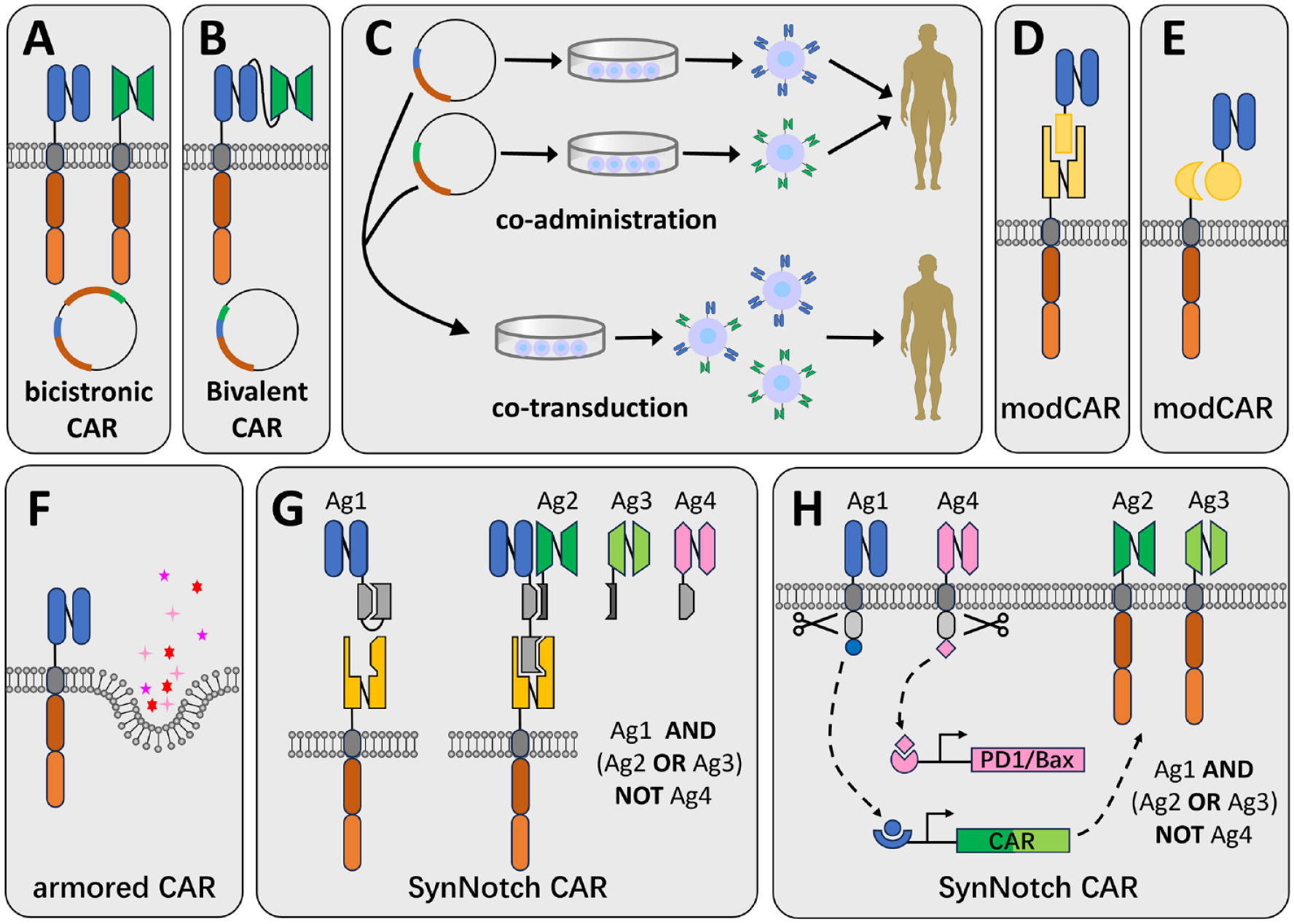
Design strategies of CARs. Schematic of CARs designed to target tumors. (A) A bicistronic CAR-engineered cell can be generated by the transduction of a bicistronic vector encoding two independent CAR molecules into a single cell. (B) A bivalent CAR is generated by transducing a vector encoding a tandom CAR molecule into a cell. (C) Bispecificity by co-administration of two different CAR-engineered immune cell products (upper) or co-transduction of two vectors encoding different CARs into immune cells (lower), but the products will be complex and expensive. A modCAR can be generated using antibodies (D) or other adaptors (E). (F) Armored CARs are designed to co-express molecules, including cytokines and/or chemokines, to increase effectiveness and durability. (G) SynNotch CARs by adding secreted proteins and binding agents, and utilizing specially designed pairing mechanisms to execute AND, NOT, and OR Boolean logic gates. This can also be achieved using specially designed transcription factors and transcription mechanisms (H). CAR: chimeric antigen receptor; modCAR: modular CAR.
In addition to the optimization of ScFv, the extracellular region has also been improved. Unlike traditional CARs that act directly on target cells, researchers have proposed a new concept: modular CAR (modCAR) 66 . This type of CAR-T (Fig. 1D, E) usually works together with its adaptors, one end of the adapter binds to the cell, and the other end is used to recognize and bind to tumor surface antigens. The adaptor molecules can be monoclonal antibodies, antibody fragments (Fig. 1D), small molecules, or any structure (Fig. 1E) that can target at least one desired antigen and mediate the crosslinking between target and effector cells. It offers a highly flexible, customizable, and universal way to rapidly optimize the effector cell activity, potentially enabling intelligent antigen targeting.
Armored CARs (Fig. 1F) are designed to secrete cytokines or other molecules that help counteract the immunosuppressive nature of the TME, potentially improving CAR-engineered cell persistence and efficacy. Sometimes referred to as the fourth-generation CARs or TRUCKs (T cells redirected for antigen-unrestricted cytokine-initiated killing), they offer a multifunctional treatment for CAR-targeted tissue that surpasses the capabilities of conventional CAR-T cells 67 . The TRUCK concept is presently being explored by co-expressing molecules like chemokine ligands or utilizing a cytokine panel based on the second-generation or third-generation CAR. Among these, IL-7, IL-12, IL-15, IL-18, IL-23, and various combinations of these molecules are in early phase trials 67 . Notably, IL-7 and IL-15 have been found to promote CAR-T cell proliferation68,69, while IL-12 and IL-18 bolster T cell effector capabilities70,71. In addition, engineered CAR-T cells expressing specific molecules, such as CCL-19 (C–C motif chemokine ligand 19) 72 or CCL-21 73 , chemotactically draw other immune cells or CAR-T cells toward nearby cancerous cells. This enhances immune cell infiltration and CAR-T cell survival, leading to increased effectiveness of CAR-T cells.
A more recent development in CAR design is synthetic notch (SynNotch) receptors (Fig. 1G, H), which offer a precise targeting strategy that allows precise control of CAR-engineered cell activation while minimizing off-target effects by incorporating an inducible switch. A typical case uses the principle of Boolean logic and logic gates 74 , co-localization-dependent protein switches (Co-LOCKR) that can execute AND, NOT, and OR Boolean logic gates are activated through conformational changes when all conditions are met. The specificity in targeting cancer cells has been demonstrated in vitro, but it still faces various challenges in clinical application.
Intracellular domains: Signaling and cell function
The intracellular domain consists of a series of co-stimulatory domains, and the classification of CARs is based on the number of co-stimulatory domains. First-generation CARs consist solely of CD3ζ, while second-generation CARs contain one co-stimulatory domain in addition to CD3ζ, and third-generation CARs involve more than one co-stimulatory domain along with CD3ζ 58 .
In T cells, first-generation CARs have been constructed containing the rearranged gene segments encoding for the variable region domains of anti-2,4,6-trinitrophenol (TNP) antibody spliced to either one of the C-region of the α or β T cell receptor (α or β TCR) chains 75 . After transducing T cells, this structure can be stably expressed on the surface of T cells and recognize tumor cells with MHC non-restriction, but it shows a weak anti-tumor effect in clinical trials 76 . The second-generation CAR-T draws on the classic signals of T cell activation and adds a co-stimulatory molecule, such as 4-1BB (also known as CD137) or CD28, which significantly improves the activation level and proliferation capability of CAR-T cells. Clinical data support that patients receiving second-generation CAR-T cell therapy have prolonged and effective control over their tumor burden64,77. Newly discovered co-stimulatory molecules presently include inducible co-stimulatory molecules (ICOS), OX40 (also known as CD134), and CD40, among others. The second-generation CAR structures are the most widely used in clinical practice. The third-generation CAR-T cells contain two co-stimulatory molecules, resulting in enhanced cytotoxicity. Longer persistence and superior expansion were observed in clinical trials when co-administering lymphoma patients with CD19-directed second-generation CAR-T cells (with CD28 co-stimulatory domain) and third-generation CAR-T cells (with CD28 and 4-1BB) 78 . However, there is still limited clinical data available for evaluating solely third-generation CAR-Ts 79 . In an investigator-initiated trial, HD-CAR-1, a third-generation CAR-T therapy, was evaluated in adult patients with relapsed/refractory ALL. At the end of the 90-day study period, 80% of patients achieved complete remission (CR) 80 . Furthermore, recent studies demonstrate that CAR-T cells equipped with both 4-1BB and CD28 co-stimulatory domains enhance the T cell memory phenotype, exhibiting better proliferation and cytokine release levels compared with those with only one co-stimulatory molecule64,81.
Most studies involving CAR-NK cells thus far have used CAR constructs originally used on T cells. Second-generation CAR-NKs with CAR constructs incorporating the CD28 82 or 4-1BB 82 co-stimulatory domain have been developed. Recent studies reported CAR-NKs with intracellular domains containing 4-1BB and CD3ζ, overcome inhibitory signals and inducing NK cell specificity, resulting in the killing of CD19+ aALL cells 83 . However, new CAR designs have been created specifically for NK cells, and these different CAR constructs have distinct effects on the cytotoxicity and cytokine production of NK cells. Research indicates that CAR-NK cells containing the NK-specific co-stimulatory domain 2B4 (also known as CD244) have significantly enhanced cytotoxic activity against tumor cells, inducing rapid proliferation, increasing cytokine production, and reducing apoptosis84,85. This indicates that NK cell-specific activation signals play a crucial role in CAR performance compared with conventional 4-1BB CARs on NK cells. Furthermore, CAR constructs utilizing different signaling domains, such as CD3ζ, DAP10, and DAP12, have demonstrated differential anti-tumor activities in primary NK cells or NK92 cell lines. Specifically, CARs with the CD3ζ signaling domain exhibited superior performance compared with those containing the DAP10 domain, while the CAR utilizing the DAP12 domain outperformed the one containing the CD3ζ domain86,87.
CAR-M cells have the same structure and generation classification as CAR-T cells, but the functions of their intracellular signaling domains are different, primarily to enhance phagocytosis. CAR-M cells have the ability of directly utilize the CD3ζ intracellular domain 88 to transmit phagocytic signals. Gamma subunit of Fc receptor (FcRγ) and multiple epidermal growth factor-like domain protein 10 (Megf10) can also be used as intracellular signaling domain88,89. A recent result indicated that the addition of a tandem PI3K recruitment domain increased the engulfment of cancer cells 89 . The first clinical trial of CAR-M is a first-generation product, containing CD3ζ as its intracellular domain 88 , and the results from phase I clinical trial showed promising initial safety and efficacy results 90 , and was granted a fast track designation by the FDA.
Improve the Efficiency of the Gene Transfer Methods
The introduction of nucleic acids in immunotherapies, including gene expression and CRISPR gene editing, presents a crucial yet challenging step91–93. In recent years, non-viral transfection techniques have become increasingly popular due to their economic benefits in overcoming the labor-intensive and expensive procedures associated with viral vector manufacturing and regulatory requirements in cGMP (Current Good Manufacturing Practice) compliance 94 . For instance, transposons or mobile genetic elements that could be utilized to achieve efficient and persistent transgene expression have gained significant interest in the generation of stable CAR-T cells95,96. In addition, electroporation or mechanoporation enables transient expression of genome-editing nucleases in immune cells, such as for the purpose of CRISPR-Cas9 expression97,98 and has been explored in mRNA-based human T cell modification 99 . These non-viral transfection techniques offer advantages in terms of procedure simplicity, capacity, and throughput, as well as a favorable safety profile94,96. Compared with non-viral transfection, viral transduction methods are the prevailing modality in clinical settings, and are widely recognized as a highly effective approach for transfecting challenging cell types100,101. Commonly used viral vectors, such as adeno-associated virus vectors and adenovirus vectors, are unable to integrate into the host genome 102 . However, retroviral vectors and lentiviral vectors can integrate, and also exhibit high delivery efficiency, making them well suited for transducing highly replicating cells, such as immune cells 103 . Tecartus 104 and Yescarta 3 , both manufactured by Kite Pharma and rely on gamma retroviral (GRV) vectors to transduce the CAR constructs into T cells, while Kymriah is transduced using a lentiviral (LV) vector 105 . In general, gamma-retroviral and LV systems are frequently utilized for transferring genes into T cells and research indicates that they typically yield 30%–80% transduction efficiency106–108. Nevertheless, safety, efficiency, and convenience concerns are concerns that must be considered 109 .
In comparison with T cells, viral gene delivery to primary NK cells has proven to be both challenging and less efficient, potentially due to the innate properties that characterize NK cells 110 . Studies demonstrated that successful transfection of primary expanded NK cells with retroviral vectors, achieving a single-round transfection efficiency range of 27%–52%111,112, and 47%–75.4% after two rounds of transfection 111 . While retroviruses have been widely used to produce CAR-NK cells in recent preclinical and clinical studies, the lack of an efficient gene transfer method for primary NK cells remains a major obstacle in the use of NK cells in immunotherapy 110 . Moreover, insertional mutagenesis and deleterious effects on primary NK cell viability associated with retroviral transfection are the major limitations in clinical applications. Compared with retroviral vectors, lentivirus-based transfection poses lower risks of genotoxicity and insertional mutagenesis 113 . Nevertheless, primary NK cells exhibit low efficiency in LV transduction, requiring multiple rounds of transfection 114 . Recently, Bari et al. 110 demonstrated that LV vectors pseudotyped with the baboon envelope glycoprotein variant (BaEV-LV) exhibit a transduction efficiency over 20 fold higher than that pseudotyped with vesicular stomatitis virus glycoprotein (VSV-G-LV), which is the first and still most widely used glycoproteins for pseudotyping LV vectors. They successfully expressed CD19-CAR in about 70% of primary human NK cells from different donors through this transfection method, and these CD19-CAR NK cells efficiently and specifically eradicated CD19-positive tumor cells. BaEV-gp pseudotyped lentivirus with a tumor-specific CAR showed an almost 100% transduction rate in NK92 cells and 50%–80% in iPSC-derived NK cells or activated primary NK cells 115 . Thus, it may be a promising vector for CAR gene transfer in NK cells. In addition, lentivirus pseudotyped with the envelope protein of gibbon ape leukemia virus (GaLV) can effectively transduce primary NK cells 116 . Electroporation and lipofection methods are also used for delivering exogenous genes in NK cells. Compared with viral transfection mentioned above, these methods offer faster transgene expression, reduced apoptosis levels, smaller inter-individual differences, and higher gene transfer efficiency 111 . However, exogenous DNA typically does not integrate into the genome of the target cells, resulting in transient expression of the transgene that declines in about 3–5 days after transfection. But when combined with DNA integration techniques, these approaches can produce stably transfected cells that express transgenes. DNA transposons are mobile DNA elements that can efficiently transpose between vectors and chromosomes via a cut-and-paste mechanism 117 . PiggyBac (PB) and Sleeping Beauty (SB) are the two most commonly used transposon systems so far, and displaying the highest transposition activity in mammalian cells when compared with other systems. By transfecting CAR-containing plasmids into iPSCs, along with transposase DNA, CAR-iPSC-NK cells were stably created, expressing CAR molecules 118 . Compared with viral vectors, transposon systems offer numerous benefits, such as lower immunogenicity, increased biosafety, reduced production costs, and the ability to transfer gene fragments with a cargo capacity approaching 9–10 kb 117 . These advantages make it a compelling choice for integrating CAR into the genome of NK cells with long-lasting expression. However, the full utilization of the transposon system for transfecting primary NK cells requires optimization, such as improving transduction efficiency and cell viability through electrotransfection.
Macrophages are more difficult to transfect and resilient to genetic manipulation than other cells in the hematopoietic system due to their capacity to recognize and respond to foreign nucleic acids 119 . Bobadilla and colleagues have created a novel HIV-1-based LV system, which has proven to be effective at delivering transgenes to bone marrow cells through induced degradation of SAMHD1 by Vpx upon infection 120 . Klichinsky’s team utilized a replication-deficient chimeric adenoviral vector, Ad5F35, to deliver CAR to macrophages efficiently and consistently, concurrently promoting the M1 phenotype, ultimately boosting tumoricidal activity. These results emphasize the potential of genetically engineered macrophages in cancer immune therapy 121 . Like methods in generating CAR-NK cells, transposition systems have been utilized to integrate genes of interest into the host genome 122 .
Novel Cell Source Methods Drive Off-the-Shelf Immune Cell Therapy Products
To date, all commercially approved cancer immune cell therapies rely on autologous use, whereby the source material used is the patient’s own cells. Although this approach has proven effective, there are important limitations, such as undesirable wait times for manufacturing when dealing with patients with rapid cancer progression, the possibility of manufacturing failure by patient’s T cell dysfunction 123 , the risk of product contamination by malignant leukemic cells extracted alongside healthy lymphocytes 124 , high production costs, complexities in standardization, and restricted opportunities for redosing. As a result, current research efforts are focused on developing allogeneic therapies to overcome the limitations of autologous therapies. Notably, CAR-NKs is a promising allogeneic adoptive cell therapy without major toxic side effects against both solid tumors and hematological malignancies 125 . It is a simpler, re-injectable, and more affordable universal treatment. However, current treatments have not demonstrated strong efficacy and/or long-term persistence, and NK cells are more difficult to engineer 126 . Research on CAR-NK cells mainly at preclinical and early-stage clinical trials, with few clinical trials yet reported 125 . Nevertheless, universal allogeneic CAR-NK cells still suggest great potential in cancer treatments 127 . Currently, CAR-M is primarily an autologous therapy, but “off-the-shelf” allogeneic CAR-M has appeared and is undergoing further study 128 .
For developing allogeneic approaches, the most significant risks must be systematically addressed: immune rejection and the development of GvHD 129 . Immune rejection can restrict the therapy’s efficacy, as the recipient’s immune system may reject the graft. GvHD can be life-threatening, caused by the endogenous αβ TCR complex of donor T cell recognition of host peptide–human leukocyte antigens (HLA) complexes, resulting in tissue damage123,130,131. To address the allogeneic risks, two main strategies can be employed. The first is performing extra genetic modifications in addition to the introduction of CAR transgenes on donor αβ T cells. Another is selecting alternative cell sources or subpopulations, such as induced pluripotent stem cells (iPSCs), umbilical cord blood (UCB) cells, γδ T cells, memory T cell subpopulations, virus-specific T cells 129 (VST), and so on 132 .
To eliminate endogenous αβ TCR-mediated GvHD, the most straightforward approach is disrupting the gene encoding for the T cell receptor constant (TRAC) 123 α chain. A case was developed by Eyquem et al. 97 to incorporate the CAR construct into the TRAC locus by adenovirus transfection and CRISPR–Cas9 knock-in technology, which induced CAR expression with TCR inactivation concurrently. Compared with random integration, CAR expression can be regulated by the endogenous TCR promoter and mimics TCR transcription when exposed to antigen, preventing constant, and excessive T cell activation, which could result in T cell differentiation and exhaustion 97 . In addition, research results demonstrated that these TRAC–CD19 CAR-T cells displayed greater anti-tumor potency in a mouse model of ALL than T cells with a retrovirally encoded CAR 97 . Furthermore, besides CRISPR-Cas9, gene-editing technologies, such as TALENs and zinc-finger nucleases (ZFNs), have also been found effective in eliminating TCR expression to create “universal” donor T cells133–137. However, while CAR-T cells with TCR deletion have reduced the risk of GvHD, their long-term persistence is shorter, possibly due to immune rejection 129 . Therefore, scientists knocked out the β2-microglobulin (B2M) gene to restrict HLA class I molecule expression 138 . This approach prevents allogeneic CAR-T cells from being recognized as foreign through their TCR, but are still vulnerable to lysis by host NK cells 98 . Therefore, a study utilizes mutant B2M fusion proteins to address this issue 139 . To increase the efficiency of HLA-II elimination during activated allogeneic CAR-T cell production, some scholars propose the inhibition of the CIITA or HLA-DRA, HLA-DQA, and HLA-DPA genes 138 .
iPSCs have unlimited proliferation and differentiation potential, making them an ideal resource for creating a repository of numerous homozygous HLA combinations 140 . By selecting an HLA-matched iPSC source from the bank, the risk of immune rejection of CAR-T cells can be minimized between the host and the graft, but stringent quality controls are essential to ensure safety, as undifferentiated, proliferating iPSCs can trigger adverse outcomes 141 . Wang et al. generated hypoimmunogenic iPSC-derived CAR-T cells that lack HLA-I/II, B2M, and CD155. The study demonstrated their efficacy in inhibiting tumor progression in vivo by exerting effector functions 142 . Furthermore, iPSCs have also paved the way for producing CAR-NK and CAR-M cells that meet clinical needs. Compared with differentiated NK cells or macrophages, iPSCs can be more efficiently engineered to stably express CARs. Li et al. 118 modified the culture medium by adding various cytokines and pro-differentiation proteins to differentiate CAR-transfected iPSCs into CAR-NK cells. These iPSC-derived CAR-NK cells exhibit potent anti-tumor activity both in vitro and in vivo 118 . Zhang et al. 128 established a bone marrow/macrophage differentiation protocol to induce the differentiation of CAR-iPS into bone marrow cell lines, successfully generated sufficient CAR-iMac cells (iPSC-derived CAR-M cells). However, the efficacy of CAR-iMacs was limited when tested in mouse models 128 . Overall, the development of iPSC-derived engineered immune cells involves intricate production procedures that necessitate exact cell product fabrication and quality assurance to bolster more extensive clinical applications.
γδ T cells express TCRγδ and recognize malignant cells in an HLA-independent manner. It does not require the elimination of TCR expression or signaling and has low or non-existent risk of GvHD143–145. Research has shown that γδ CAR-T cells exhibit similar or greater cytotoxic effects than conventional αβ CAR-T cells with no GvHD observed in vivo. However, their cytotoxic capacity in vitro was less persistent143,146. In addition, the anti-CD20 γδ CAR-T cells resulted in promising outcomes in animal models of B cell lymphoma and are currently undergoing phase I clinical trials 143 . Notably, TCRs targeting a wide variety of tumors in an HLA-independent way and derived from both αβ and γδ TCR repertoires have been identified, allowing for the engineering of immune cells that attack tumor-specific antigens commonly shared among patients 147 .
UCB provides an easy source of NK cells and allogeneic T cells. They are enriched with γδ T cells and hematopoietic stem cells (HSCs), even though the total cell number is limited148,149. UCB-derived CAR-T cells exhibited enhanced tumor suppression in vivo, reduced IL-10 secretion, and a lower proportion of regulatory T cells (Tregs), indicating improved therapeutic potential 150 . Van Caeneghem et al. isolated CD34+ HSCs from cord blood and then generated CAR-T cells. These CAR-T cells have a naive phenotype (CD45RA+ CD62L+) and do not express TCRαβ complexes, which significantly reduced the risk GvHD and eliminate tumor cells effectively 149 .
Memory T cells, characterized by CD45RA−CD45RO+ phenotype 151 , are less likely to develop GvHD and less likely to migrate to organs that manifest GvHD 152 . Studies have indicated that T memory stem cells (TSCM) and central memory T cells (Tcm) possess superior proliferative and stemness potential compared with their differentiated counterparts 153 and have exhibited high expansion and persistence in clinical studies following adoptive T cell transfer154,155.
VSTs possess a TCR capable of recognizing viral antigens, such as cytomegalovirus (CMV), Epstein–Barr virus (EBV), and others. VSTs that have been modified with CARs have exhibited promising results, particularly in solid tumors156,157,158, but engineering tumor-targeted transgenic TCR VSTs are more challenging than CARs147,159. In patients with Hodgkin lymphoma expressing EBV-associated antigens in their tumor cells, infusion of CAR-VST cells has yielded good tolerance and remission rates. A HER2-targeted CAR-VST cell product was shown to be both safe and clinically effective for patients with CMV seropositivity and progressive glioblastoma in a phase I clinical trial 157 . Notably, VST banks can be established to cover majority of patients with an appropriately matched line and facilitate the redirection of CAR-T or TCR-T to tumors for clinical use 160 .
PBMCs are typically obtained from the peripheral circulation or cord blood and so on. It is an important source of primary NK cells for clinical purposes. PBMC-derived NK (PB-NK) cells can be isolated from both HLA-matched and mismatched donors, expanding the pool of potential donors. Typically, they display a mature phenotype, reduced proliferative capacity, and increased cytotoxicity, without the risk of GvHD115,161. Töpfer et al. 87 have demonstrated that CAR-expressing NK cell line YTS, as well as PB-NK cells, can be successfully redirected against prostate stem cell antigen (PSCA)-positive tumors, leading to complete tumor eradication in a significant fraction of treated mice. A transiently CAR-PBMC cell product expressing a mesothelin-targeting mRNA CAR, MCY-M11, has entered into clinical trials (NCT03608618), it is a mixed product containing diverse CAR-engineered T cell subsets, monocytes, and B cell populations, exhibits desirable functional and immune phenotypic characteristics 162 .
NKT cells also called CD1d-restricted T cells, are a heterogeneous group of T cells that possess both T and NK cell characteristics. These cells predominantly identify the non-polymorphic CD1d molecule, an antigen-presenting molecule that binds to both self and foreign lipids, and glycolipids. Type I NKT or iNKT cells lack HLA-restriction and may function as universal donor cells, but the persistence in vivo remains unknown. Research indicates that CAR19-iNKT cells exhibit anti-lymphoma activity and an enhanced capacity to eliminate brain lymphomas 163 . Another allogeneic NKT, expressing anti-CD19 chimeric receptor with CD28 and IL-15, is currently undergoing clinical trials for patients with relapsed or refractory B cell malignancies (NCT03774654).
Modify the Tumor Environment for Therapeutic Immune Cells
Low persistence and inadequate proliferation following infusion are significant challenges to the effectiveness of immune cell therapy for solid tumors, largely attributed to an immunosuppressive microenvironment, which acts as an active promoter of cancer progression, rather than a passive bystander 164 . Tumor cells inhabit this heterogeneous microenvironment composed of infiltrating and resident host cells (such as T cells and macrophages), secreted factors [such as IL-10 and transforming growth factor-beta (TGFβ)], and extracellular matrix (ECM) 164 . These components can interact to induce a supportive environment for malignant cell growth, migration, and metastasis, thereby evading the immune system and tumor-specific Tc cells165,166.
To eradicate tumors, scientists have developed several strategies to promote the migration and infiltration of CAR-engineered or TCR-engineered therapeutic immune cells to cancer focus, including the utilization of chemokine receptors like CCR2167,168, CXCR2169,170, CCR4 171 , CXCR4 172 , and others. These studies demonstrate that CAR-T, TCR-T, and NK cells exhibit increased infiltration rates into tumors, resulting in tumor regression and enhanced anti-tumor efficacy.
However, infiltrating genetically engineered immune cells into tumors is only the first step in fighting cancer. Sometimes, direct transformation methods may be necessary to modify the immunosuppressive microenvironment. Utilization of immune checkpoint inhibitors, such as anti-PD-1, anti-PD-L1 antibodies, and anti-CTLA4 antibodies, can ameliorate the immunosuppressive TME 173 . However, it can lead to the emergence of drug resistance, T cell exhaustion and relapse, and sometimes even life-threatening toxicities174,175. Blocking TGF-β signaling in the TME is an alternative approach to improve checkpoint inhibitor efficacy and enhance anti-tumor responses, due to its crucial role in tumor signaling, remodeling, and metabolism. Clinical evidence also demonstrates a correlation between TGF-β signaling and non-responsive patients to checkpoint blockers. To address this, bifunctional antibody-ligand traps have been developed 176 using an antibody that targets CTLA-4 or PD-L1 fused with a TGFβ receptor II ectodomain (TGFβRIIecd). It can sequester TGF-β within the TME, deplete Tregs, and facilitate the CTL co-stimulation. In addition, TGFβ-resistant tumor-specific T cells have been developed through the transduction of a dominant-negative TGFβ receptor-II (dnTGFβRII), and have demonstrated therapeutic benefits in animal models and clinical studies177,178.
Another novel strategy is to take advantage of the high concentration of immunosuppressive signaling molecules in the TME by introducing a chimeric switch receptor (CSR), such as PD-1: CD28179–182, TIGIT:CD28 183 , CTLA-4:CD28 184 , TGF-βRII:4-1BB 185 , Fas:4-1BB 186 . A CSR comprises a PD-1, CTLA-4, or TGF-βRII extracellular domain and a CD28 or 4-1BB intracellular domain. T cells were transduced with the CSR together with a CAR179,180 or TC181,182 to generate engineered CTLs. These engineered CTLs still interact with immunosuppressive molecules on tumor cells, but deliver co-stimulation signals via the intracellular domain instead of an inhibitory signal. Recently, this strategy has undergone clinical testing. These engineered cells are found to effectively overcome the immunosuppressive TME, like secrete more IFN-γ, accumulating in tumors, and exhibiting superior anti-tumor functionalities 173 .
The ECM creates a physical barrier for various anti-cancer therapies. However, macrophages play a crucial role in regulating the ECM and exhibit tumor-homing behavior 187 . CAR-engineered macrophages were shown to actively migrate to tumor sites, could locally deliver cytokines and/or cytotoxic substances to antigen-specific environments when used as a drug delivery system, significantly alter the immune-suppressive TME and eliminate tumor cells through phagocytosis187,188. Zhang et al. 189 used CAR-modified macrophages to solve the problem of insufficient immune cell infiltration into tumors caused by the ECM. In vitro studies have demonstrated that CAR-147 macrophages are capable of remodeling the ECM of tumors, inducing CD3+ T cell infiltration, and elevating the levels of IL-12 and IFN-γ in tumor niches, ultimately exhibiting an anti-tumor effect 189 .
Clinical Research Progress
From the perspective of clinical progress, 10 CAR-T products have been approved worldwide since the FDA approved the first CAR-T cell therapy products in 2017 (Supplementary Table 1). The clinical research of CAR-T, CAR-NK, and TCR-T therapies has been rapidly increasing since 2013, with the number of CAR-T cell therapies reaching 258 cases in 2022, it should be noted that the data for 2023 are from January to October (Fig. 2A). From a national perspective, clinical research on high-risk cell therapies is mainly distributed in the United States and China, accounting for more than 80% of the world’s research (Fig. 2B). The main clinical treatment targets of CAR-T therapy are CD19, CD22, CD20, and B-cell maturation antigen (BCMA). Among blood tumors, the fastest-growing indications are lymphoma and myeloma.
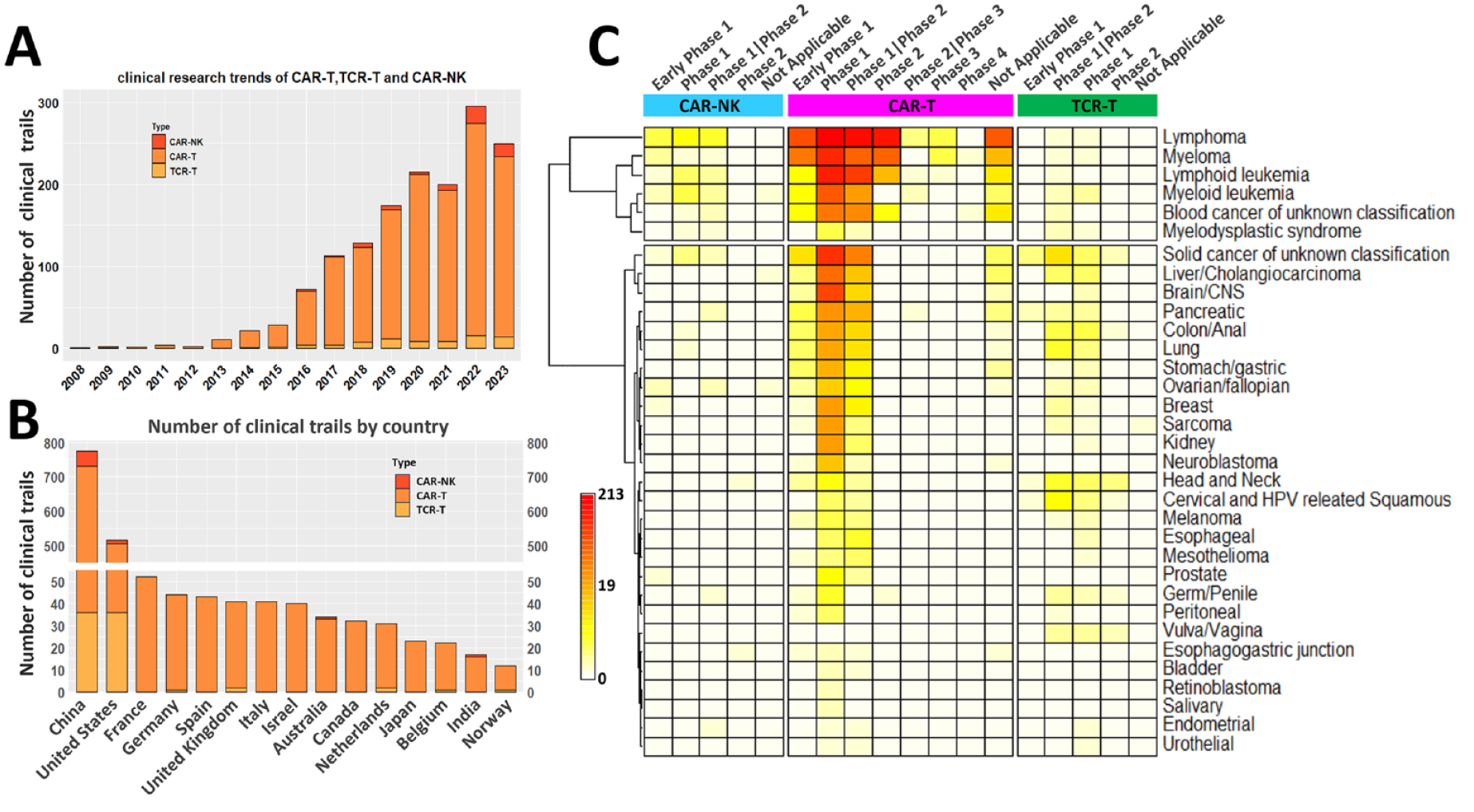
CAR-T, TCR-T, and CAR-NK therapies on clinical trials. Clinical trial data statistics. (A) Number of cell therapy clinical studies from 2018 to 2023, grouped as CAR-T, TCR-T, and CAR-NK. (B) Number of cell therapy clinical studies by countries, grouped as CAR-T, TCR-T, and CAR-NK, the Y-axis was broke off between 50 and 450. (C) Clinical progress of cell therapy drugs regarding indications, the cell therapy drugs were grouped the same as (A), and the indications were grouped as liquid and solid tumors. Raw data downloads from https://classic.clinicaltrials.gov/, updated to October 30, 2023. CAR-T: chimeric antigen receptor-T cell; CAR-NK: chimeric antigen receptor-natural killer; CNS: central nervous system; HPV: human papilloma virus; TCR-T: T cell receptor-T.
Currently, TCR-T has one therapeutic product on the market, which is used for metastatic uveal melanoma 190 . In general, TCR-T tends to progress faster in solid tumors than in hematological tumors (Fig. 2C). The target antigens of TCR-T are mainly concentrated in MART-1, HPV16-E6, NYESO, HPV16-E7, and so on 191 . In terms of conditions, the clinical research on TCR-T is mainly used in the treatment of solid tumors. Most of these solid tumors are related to viral infections, such as cervical cancer, which is associated with human papilloma virus (HPV) infection. In addition, clinical research of TCR-T on cancers located in the head and neck, liver, and colon are progressing rapidly (Fig. 2C). In terms of CAR-NK conditions, hematological tumors are still the fastest progressing, among which lymphoma conditions progress faster (Fig. 2C). The clinical treatment targets of CAR-NK are mainly CD19, CD22, and BCMA for blood tumors and PSMA, HER2, ROBO1, and MUC1 for solid tumors 191 . Overall, TCR-T and CAR-NK are still in early clinical development. So far, clinical research on CAR-M is still in its infancy, with only one phase I clinical trial (NCT04660929).
The data of Fig. 2 are available from Supplementary Table 2.
Conclusion and Perspectives
Immune cell therapy is becoming increasingly important in the treatment of tumors. In contrast to alternative immunotherapeutic modalities, such as immune checkpoint inhibitors 192 and cytokines 193 , immune cell therapy has exhibited superior efficacy, specificity, and propensity for inducing acceptable side effects. When used as a standalone treatment for certain cancers, immune cell therapy, specifically adoptive T cell transfer therapy has shown remarkable success in hematologic malignancies, such as leukemia and lymphoma. However, some solid tumors have proven more challenging for immune cell therapy alone due to factors, such as the immunosuppressive TME, tumor heterogeneity, low expression of target antigens, and so on. In many cases, switching to another immune cell therapy 194 or combining immune cell therapy with other therapeutic strategies is considered beneficial, such as chemotherapy, radiation therapy, other immunotherapy, or targeted therapy, and may enhance overall treatment efficacy through distinct mechanisms195,196. Currently, there are 10 CAR-T products approved worldwide, while the DC vaccine and TCR-T have only one approved product. In the aspect of low-risk immune cell therapy, both TILs and DC vaccines show promise in treating solid tumors. In addition, CIK therapy suggests synergistic anti-tumor effects in combination with other immunologic strategies. These methods typically consist of heterogeneous cells with varied functional characteristics, offering advantages in treating malignancies with relatively high heterogeneity. On the other hand, high-risk immune cell therapies, such as CAR-T and TCR-T are typically target-oriented and highly specific, making them particularly suitable for relatively rapidly proliferating malignant. In hematological tumors, CAR-T and TCR-T have been clinically verified, and their use in treating solid tumors is progressing steadily. While CAR-NK and CAR-M, based on their biological nature, have inherent advantages in the treatment of solid tumors. TCR-T focuses on specific TAA and neoantigens, making them beneficial in foreign antigens, such as virus-related malignancies. Researchers are currently working to break through the bottlenecks of cell therapy technologies including CAR-T. New cell sources and methods that reduce the risk of GvHD and other risks create a window for “off-the-shelf” or allogeneic cell therapies, that can be prepared in advance and easily administered to multiple patients when needed, enabling faster diagnosis and cancer treatment. Furthermore, the novel design of targeted CARs minimizes the damage to healthy tissues and reduces side effects compared with traditional treatments. Certain designs even offer possibilities for treating solid tumors. In addition, the strategies of modifying the tumor environment and inducing memory T cells have made it possible to achieve long-term effects 197 .
At the same time, due to their novel, intricate, and technical nature, these therapies may pose previously unexplored risks to public health and individual patients. TIL cell therapy, DC/CIK cell therapy, and DC vaccines are considered low to moderate risk. The risk factors include biologically active substances used in manufacturing, such as antibodies, cytokines, sera, growth factors, and antibiotics, along with the product stability and viability risks during preservation, freezing, thawing, and cold chain transportation. In addition, the products themselves may carry inherent risks, such as incomplete removal of tumor cells or other unwanted cells, as well as potential complications or reduced product activity associated with homing, grafting, migration, and proliferation. However, high-risk immune therapies necessitate more manipulation for the use of either genetically modified cells, autologous stem cells, or allogeneic or xenogeneic cells, which require additional evaluation. Clinically, these therapies can sometimes trigger cytokine storm and cytokine release syndrome (CRS), both of which are life-threatening systemic inflammatory syndromes. Therefore, the risk management considerations for these products are correspondingly more challenging. The potential risks of both the immediate and delayed impacts arising from the interaction between the product and the patient must be carefully evaluated. These risks encompass unwanted immunogenicity and its associated effects, including CRS, GVHD, graft rejection, anaphylaxis, hypersensitivity reactions, and immune deficiencies. In addition, it is important to evaluate the potential risks associated with both intended and unintended genetic modification of the patient’s cells, including alterations in cellular function, cell apoptosis, changes in growth or differentiation, insertional mutagenesis or genotoxicity, as well as the possibility of malignancy.
However, accurately assessing the risk of immune cell therapy remains challenging. Several genuine risks have exceeded expectations or were not revealed by preclinical studies. It is important to understand that risk is ubiquitous in clinical research and it is defined as the likelihood of an adverse outcome occurring within a specified timeframe 198 . Assessing an acceptable level of immune cell therapy risk involves weighing various factors beyond absolute risk against the potential treatment benefits that may ensue. Compared with patients with less severe tumors or cancer indications existing appropriate therapies, the acceptable risk levels of patients with life-threatening terminal or severe malignancies are much higher.
Overall, the technical barriers to immune cell therapy are relatively high, with a strong driving force for innovation and multiple technological developments. Progress has shown explosive growth in recent years. Despite the approval of CAR-T, TCR-T, and DC vaccines, other immune cell therapies are still in the early clinical and preclinical research stages. Low-risk therapies, such as TIL, are faced with limited effectiveness due to the inability to complete eliminate Tregs 199 . CIK cell therapy encounters difficulties in both acquisition and expansion. DC tumor vaccines are hindered by the lack of ideal tumor-specific antigens, weak TAA immunogenicity, difficulty in efficiently, inducing immunogenic DC, and the in vitro DC sensitization method of intravenous reinfusion cannot effectively activate DC differentiation and maturation. High-risk cell therapies still have a long way to go in overcoming the barriers of tumor heterogeneity. CAR-T cells are still facing challenges, such as exhaustion of effector T cells, cytokine storm, and immunosuppressive microenvironment. Challenges of TCR-T cell therapy include how to reduce TCR mismatches and minimal options for targeting neoantigens. CAR-NK therapies are experiencing genetic manipulation and cell proliferation difficulties. Although CAR-Ms could become a promising anti-cancer therapy, they have yet to be fully realized. Clinical research needs to confirm the highly adaptive nature of tumor-associated macrophages in determining whether the TME can convert tumor-targeted CAR-M into a tumor-supportive phenotype. Nevertheless, immune cell therapy still holds paramount potential in future development of anti-cancer therapies. It is widely believed that rapid technological innovations will establish these therapies as highly effective cancer treatments, significantly improving treatment outcomes and advancing human health care.
Supplemental Material
sj-xls-1-cll-10.1177_09636897241231892 – Supplemental material for The Progress and Prospects of Immune Cell Therapy for the Treatment of Cancer
Supplemental material, sj-xls-1-cll-10.1177_09636897241231892 for The Progress and Prospects of Immune Cell Therapy for the Treatment of Cancer by Jia Han, Bowen Zhang, Senyu Zheng, Yuan Jiang, Xiaopeng Zhang and Kaiyun Mao in Cell Transplantation
Supplemental Material
sj-xls-2-cll-10.1177_09636897241231892 – Supplemental material for The Progress and Prospects of Immune Cell Therapy for the Treatment of Cancer
Supplemental material, sj-xls-2-cll-10.1177_09636897241231892 for The Progress and Prospects of Immune Cell Therapy for the Treatment of Cancer by Jia Han, Bowen Zhang, Senyu Zheng, Yuan Jiang, Xiaopeng Zhang and Kaiyun Mao in Cell Transplantation
Footnotes
Author Contributions
First author and corresponding author Jia Han led the project and conceived the manuscript, wrote, and revised the entire manuscript. She also curated, analyzed, and visualized the data. Bowen Zhang, Senyu Zheng, and Yuan Jiang participated in writing the section on low-risk immune cell therapy, and Yuan Jiang participated in writing the induction. Xiaopeng Zhang optimized and arranged the pictures. Corresponding author Kaiyun Mao was in charge of expert consultation, resources, and project management. All authors have read and agreed to publish this manuscript.
Ethical Approval
There are no animal and human subjects in this article and the ethical procedure is not applicable.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the 2023 Shanghai “Technology Innovation Action Plan” Soft Science Research Project “Research on the development trends of underlying technologies for gene therapy” (No. 23692102702); and the 2022 Shanghai “Technology Innovation Action Plan” Soft Science Research Project “Research and policy recommendations on the development of cell therapy and gene therapy industries in Shanghai” (No. 22692116400).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.