
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a severe hyperinflammatory disease characterized by familial and acquired forms. Here, we present the case of a 26-year-old male patient with relapsed/refractory peripheral T-cell lymphoma and concurrent HLH. Whole-exon sequencing revealed germline mutations associated with HLH, including those in critical genes such as CD27 and UNC13D and other germline heterozygous variants (NOTCH2, NOTCH3, IL2RA, TYK2, AGL, CFD, and F13A1). CD107a analyses consistently demonstrated impaired degranulation of cytotoxic T-lymphocytes and natural killer (NK) cells. Examination of the patient’s family pedigree revealed that his father and mother harbored UNC13D and CD27 mutations, respectively; his brother carried the same CD27 heterozygous mutation. However, none of them manifested the disease. Despite the missense mutation of CD27 (c.779C>T; p.Pro260Leu) lacking previous documentation in databases, comprehensive analysis suggested non-pathogenic mutations in the CD27 variant, indicating minimal impact on T- and NK-cell functions. These results ultimately supported the option of hematopoietic stem cell transplantation (HSCT) as a successful curative therapeutic approach. As of this report, the patient has remained free of lymphoma and quiescent HLH 15.2 months post-HSCT. This study underscores the efficacy of genetic tests in identifying significant mutations and confirming their etiologies, providing an early basis for treatment decisions and the selection of suitable transplant donors.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a devastating hyperinflammatory immune disorder characterized by aberrant activation of macrophages and cytotoxic T cells, leading to hypersecretion of proinflammatory cytokines, tissue infiltration, hemophagocytosis, and organ damage 1 . HLH can be divided into two subgroups: primary HLH (pHLH) and secondary HLH (sHLH). pHLH often occurred in pediatrics with either inherited genetic mutations (e.g., PRF1, UNC13D, STX11, STXBP2, and CD27), which impair the exocytosis of cytoplasmic granules and perforin-mediated cytotoxicity, or mutations associated with primary immune deficiency/deregulatory states, such as SH2D1A, XIAP, RAB27A, and LYST2,3. In contrast, secondary HLH (sHLH) predominantly occurs in adults or older children without a demonstrable genetic driver, typically triggered by underlying hematological malignancies, infections, and autoimmune diseases 4 . The mortality in cases of sHLH can reach 80% when patients advance to the critical stage 5 . Accumulating evidence suggests that about 25% of adult-onset HLH cases have an underlying genetic predisposition within the context of a precipitating etiology, resulting in chronic immune stimulation with dysregulated regulatory feedback loops6,7. Lymphoma, particularly T- and natural killer (NK-cell lymphomas), is the most common neoplastic trigger of HLH 4 . Allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains the sole curative therapy for relapsed/refractory (R/R) HLH, especially in cases of genetic defects. Thus, determining genetic alterations and cytotoxic functions in eligible donors is imperative for optimizing transplant therapeutic effects.
Previous studies have reported a correlation between the dysregulation of the host immune response and the occurrence and progression of secondary hemophagocytic lymphohistiocytosis (sHLH)4,8. In addition, the dysfunction of cytotoxic T-lymphocytes (CTL) and NK cells has been identified as a critical factor in the pathogenesis of sHLH9,10, which contributes to an inability to clear antigenic stimulation, resulting in persistent and amplified immune responses. This, in turn, leads to the secretion of large amounts of proinflammatory cytokines by activated macrophages and lymphocytes. HLH-associated UNC13D gene-encoding protein is crucial for the cytotoxic activity of T-lymphocytes, regulating the early phase of cytotoxic particle exocytosis and subsequent vesicle docking to the cytomembrane. This mutation can lead to defects in cytotoxic granule exocytosis, resulting in the diminished cytotoxic activity of T-lymphocytes and, consequently, the occurrence of HLH 11 .
In this study, we describe a case of peripheral T-cell lymphoma and HLH complicated by multiple germline heterozygous mutations, including HLH-specific variants (UNC13D and CD27). Notably, missense mutations in CD27 have not yet been reported. CD107a analyses revealed markedly impaired degranulation of CTL and NK cells. Collectively, the analyses of the novel CD27 variant indicated a non-pathogenic mutation of CD27, providing a rationale for donor selection and supporting HSCT as a successful curative treatment approach.
Methods
Whole-Exome Sequencing
Whole-exome sequencing (WES) was performed as previously described12–15. Blood samples from patients and his family members were collected, and genomic DNA was extracted using a GenMagBio Genomic DNA Purification kit based on the manufacturer’s instructions. The DNA libraries were generated using VAHTS Universal DNA Library Prep Kit for Illumina. Twist Human Comprehensive Exome was used for capturing sequences. Then, the enriched library was sequenced on an Illumina Next500 platform. Next, WES reads were aligned to the GRCh37.p13 by using Burrows-Wheeler Aligner software (version 0.7.15). GATK Indel Realigner was then used for local realignment of the Burrows-Wheeler aligned reads. Then, the base quality recalibration of the Burrows-Wheeler aligned reads was performed by using GATK BaseRecalibrator. Next, to identify putative mutations, variant calling was performed using SAMtools (v1.5) in combination with VarScan2 (v2.3.9). Finally, all variants were annotated within Annovar software (http://www.openbioinformatics.org/annovar).
Identified variants by WES were selected for data interpretation with minor allele frequency less than 0.05 in dbSNP, GnomAD, ExAC, ESP6500, 1000 Genomes Project and BGI database for 50,000 Chinese Han samples. Based on the variant interpretation guidelines of American College of Medical Genetics and Genomics (ACMG), data interpretation was performed 16 . Finally, we compared the variants found in patient and his unaffected family members with the references of OMIM database and published literature.
Sanger Sequencing
To validate putative mutations identified in the patient, Sanger sequencing was used. Primers flanking the candidate loci were designed according to the reference genomic sequences of the Human Genome from GenBank in NCBI and synthesized by Youkang, Hangzhou, China. Polymerase chain reaction (PCR) amplification was conducted in an Eppendorf MasterCycler gradient PCR machine, and the products were subsequently sequenced on an ABI PRISM 3730 automated sequencer (Applied Biosystems, Foster City, CA, USA). Sequence data analyses were performed by SanpGene Viewer. The primers list of heterozygous variants found in WES is summarized in Supplemental Table S1.
Case Presentation
A 26-year-old male patient with a persistent fever for 2 months was referred to our department in November 2021, with no personal or family history of unexplained fever, cancer, or other diseases. Bone marrow (BM) aspiration and biopsy revealed hemophagocytosis (Supplemental Fig. S1A). Cytogenetic analysis revealed a normal 46, XY karyotype. On admission, he exhibited a body temperature of 40°C. Laboratory findings revealed progressive pancytopenia, hyperferritinemia, elevated aspartate aminotransferase and alanine aminotransferase levels, and positive Epstein–Barr virus (EBV) DNA at 2.6 × 105 copy/ml. Triglyceride, fibrinogen, and soluble CD25 levels were within normal levels. HLH functional studies indicated impaired degranulation of NK (Δ CD107a 0%) and CTL (Δ MFI 2.1) in CD107a analysis (Fig. 1A), along with normal NK-cell activity (18.66%) and perforin expression in NK and CTL. Positron emission tomography/computed tomography (PET/CT) unveiled generalized and multiple enlarged lymphoma nodes, coupled with hypermetabolism and splenomegaly, indicative of lymphoma infiltration (Fig. 1B). Further BM biopsy confirmed EBV-positive T-lymphoma (Supplemental Fig. S1B). Biopsy of the lymphoma node was not performed because of severe pancytopenia. Despite the insufficient pathological findings, the diagnosis of T-lymphoma (stage IV, group B) complicated by HLH was considered. Ruxolitinib (Rux, 10 mg) and dexamethasone (Dex) were initiated to alleviate HLH symptoms, supplemented with antiviral therapy (acyclovir plus foscarnet) to manage the EBV infection. Two cycles of E-POCH (cyclophosphamide, doxorubicin, etoposide, vincristine, and Dex) chemotherapy were then administered. After two cycles of the chemotherapy, while the biochemical indicators of HLH decreased to an average, the blood EBV copy numbers remained high. Pseudomonas aeruginosa pneumonia occurred at the end of the chemotherapy, and the patient’s condition was generally poor. He was then switched to cyclophosphamide, doxorubicin, vincristine, and prednisolone (CHOP) chemotherapy. However, on day 10 post-chemotherapy, a suspected recurrence of HLH was observed, manifesting a repeated fever, pancytopenia, hyperferritinemia (2357 ng/ml), and positive EBV-DNA. Treatment response evaluated by PET/CT showed stable disease. An inguinal lymph node biopsy was performed and confirmed peripheral T-cell lymphoma not otherwise specified (PTCL-NOS) with immunohistochemical markers that were positive for CD45, CD3, CD2, CD4, CD7, CD43, EBER, Ki-67 (40%), and PD-1, but negative for CD20, CD10, CD56, CD21, CD15, and PAX-5 (Supplemental Fig. S1C). WES of pathological BM samples detected multiple germline gene mutations, including NOTCH2, NOTCH3, IL2RA, TYK2, AGL, F13A1, CFD, CD27, and UNC13D, which were further confirmed by Sanger sequencing (Fig. 2A and Supplemental Table S2). Although UNC13D and CD27 are known HLH-predisposing genes, a novel germline missense mutation was detected in CD27 (c.779C>T, p.Pro260Leu) (Fig. 2B). Intensified HLH therapy with 15 mg/day of Rux plus Dex led to symptoms relief within 3 days, and the biochemical markers of HLH were generally relieved. A fourth cycle of CHOP plus pegaspargase was administered. Nevertheless, after 19 days of chemotherapy, the patient experienced a recurring high-grade fever, attributed to an uncontrolled lymphoma and recurrent HLH. The gemcitabine, dexamethasone, and cisplatin (GDP) regimen was subsequently employed, and chidamide (20 mg twice weekly for 3 weeks) was administered. On day 22, after GDP chemotherapy, a repeated fever was observed, leading to the administration of the etoposide, methylprednisolone, cytarabine, and cisplatin (E-SHAP) regimen. A follow-up PET/CT examination after chemotherapy revealed generally ameliorated lesions, indicative of partial remission (PR) of lymphoma (Fig. 1B). Sintilimab, a PD-1 inhibitor, was administered at 200 mg on d0 combined with the E-POCH regimen. However, this course was complicated by the development of hypovolemic shock. Subsequently, he received one cycle of the E-POCH regimen and achieved PR, recommended for allo-HSCT. The detailed treatment course is summarized in Fig. 1C.
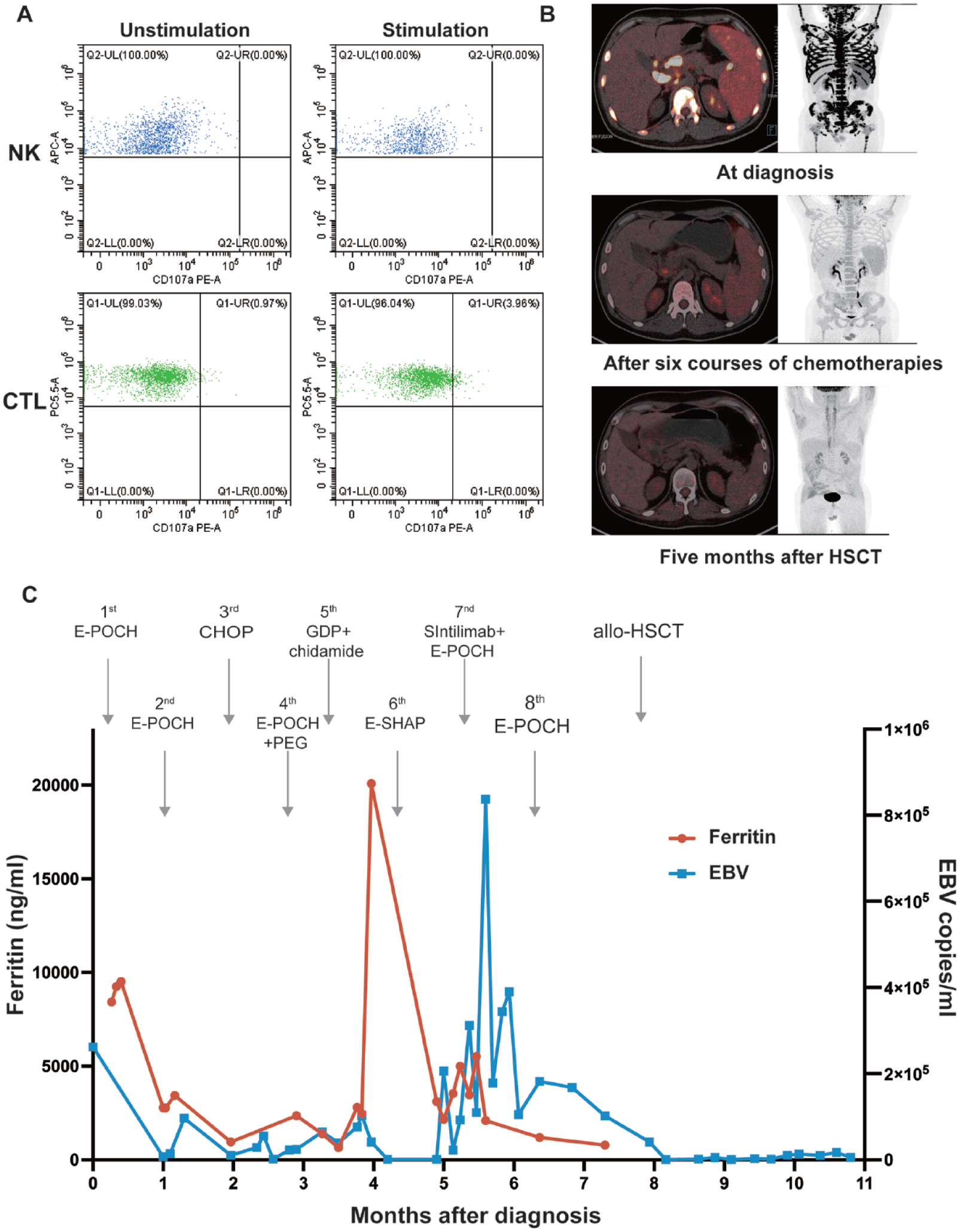
(A) Degranulation of CD56+ natural killer (NK) cells and CD8+ CTL in the CD107a analysis. CD107a expression on cell surfaces was analyzed using flow cytometry in resting cells (left dot plot diagrams) and subsequent to 48 h stimulation with IL-2 of NK cells, and phytohemagglutinin (PHA)/IL-2 of CD8+ T cells (right dot plot diagrams), respectively. (B) Clinical evaluation by positron emission tomography/computed tomography imaging. (C) Ferritin levels and Epstein–Barr virus-DNA copies throughout treatment courses. APC-A: allophycocyanin-area; CHOP: cyclophosphamide, doxorubicin, vincristine, and prednisolone; CTL: cytotoxic T-lymphocytes; EBV: epstein–Barr virus; E-POCH: etoposide, prednisolone, vincristine, cyclophosphamide, doxorubicin; E-SHAP: etoposide, methylprednisolone, cytarabine, and cisplatin; GDP: gemcitabine, dexamethasone, and cisplatin; HSCT: hematopoietic stem cell transplantation; IL-2: interleukin 2; NK: natural killer; PE-A: phycoerythrin-area; PEG: pegaspargase.
To identify suitable donors, his family members underwent WES. His father and mother were found to harbor UNC13D and CD27 mutations, respectively, and his older brother (HLA-identical, 10/10) carried part of the germline mutations inherited from his parents, including the same CD27 missense mutation (Fig. 2C and Supplemental Table S2). His brother was also evaluated by HLH-related tests. The novel CD27 missense mutation, which potentially affects T-cell function, occurs within the intracellular domain of the CD27 protein and has not yet been documented in the dbSNP database. Pathogenicity prediction algorithms offer conflicting assessments of the p.P260L variant: SIFT indicates deleterious mutations, whereas Polyphen2, LRT, and MutationAssessor suggest benign mutation. To provide further insights, we used AlphaFold 2 to speculate on the structure of this mutation, revealing a predicted structure closely resembling the wild-type structure (Fig. 3A). Flow cytometry demonstrated that the expression of CD27 was similar to that in the normal control (Fig. 3B). Moreover, NK-cell activity and granzyme B and perforin expression levels in his brother were all within normal ranges (Fig. 3C–E). Based on the aforementioned results, we hypothesized that the germline CD27 variant does not adversely affect the T-cell function, making his brother a suitable candidate for allo-HSCT.
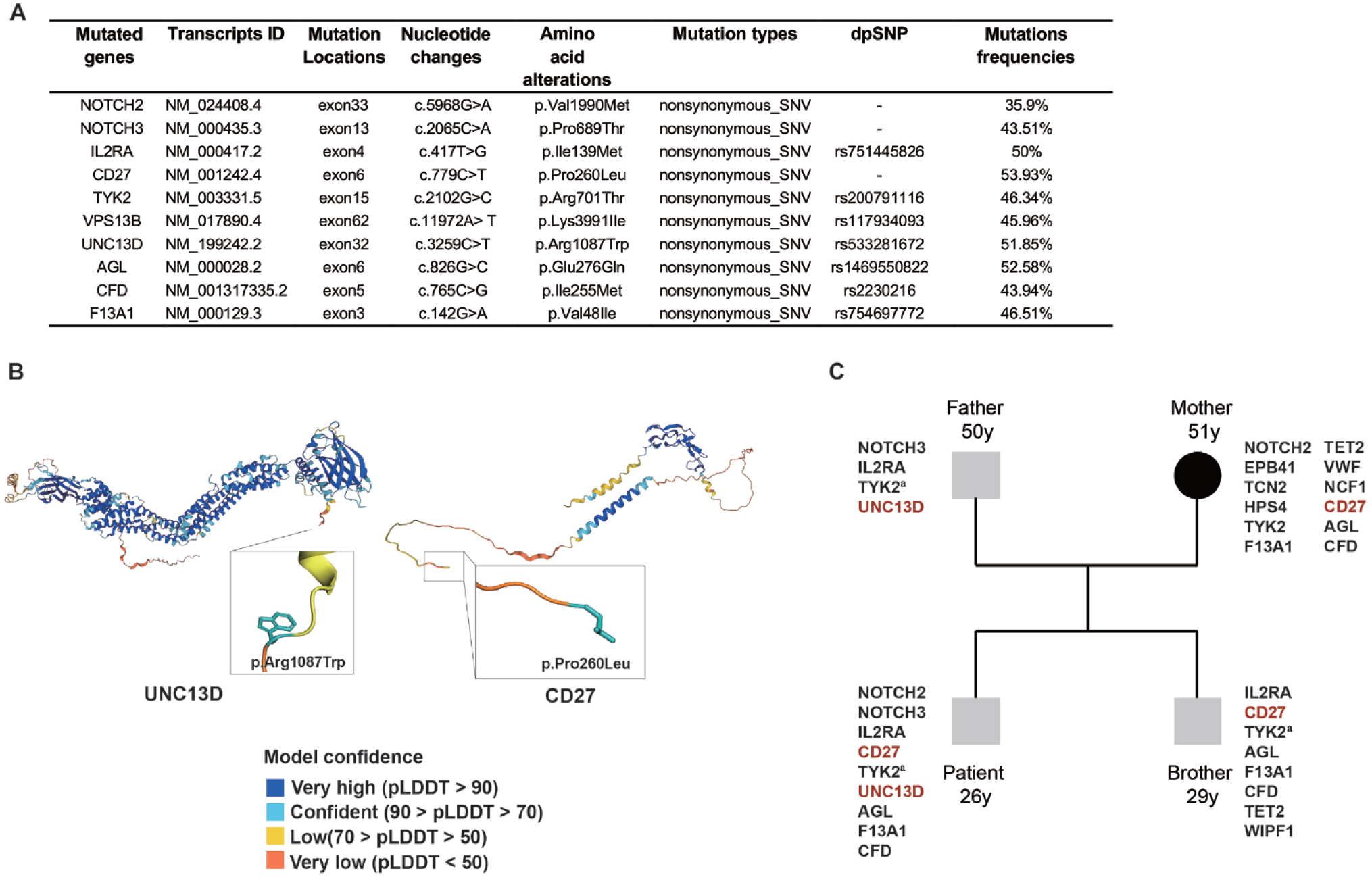
(A) Specific information on the mutated genes detected by the whole-exon sequencing. (B) Structure of the human UNC13D (left panel) and CD27 (right panel) domain highlighting the primary mutated sites. (C) Whole-exon sequencing pedigree analysis of the patient and his family members. dbSNP: The Single Nucleotide Polymorphism Database; pLDDT: per-residue Local Distance Difference Test.
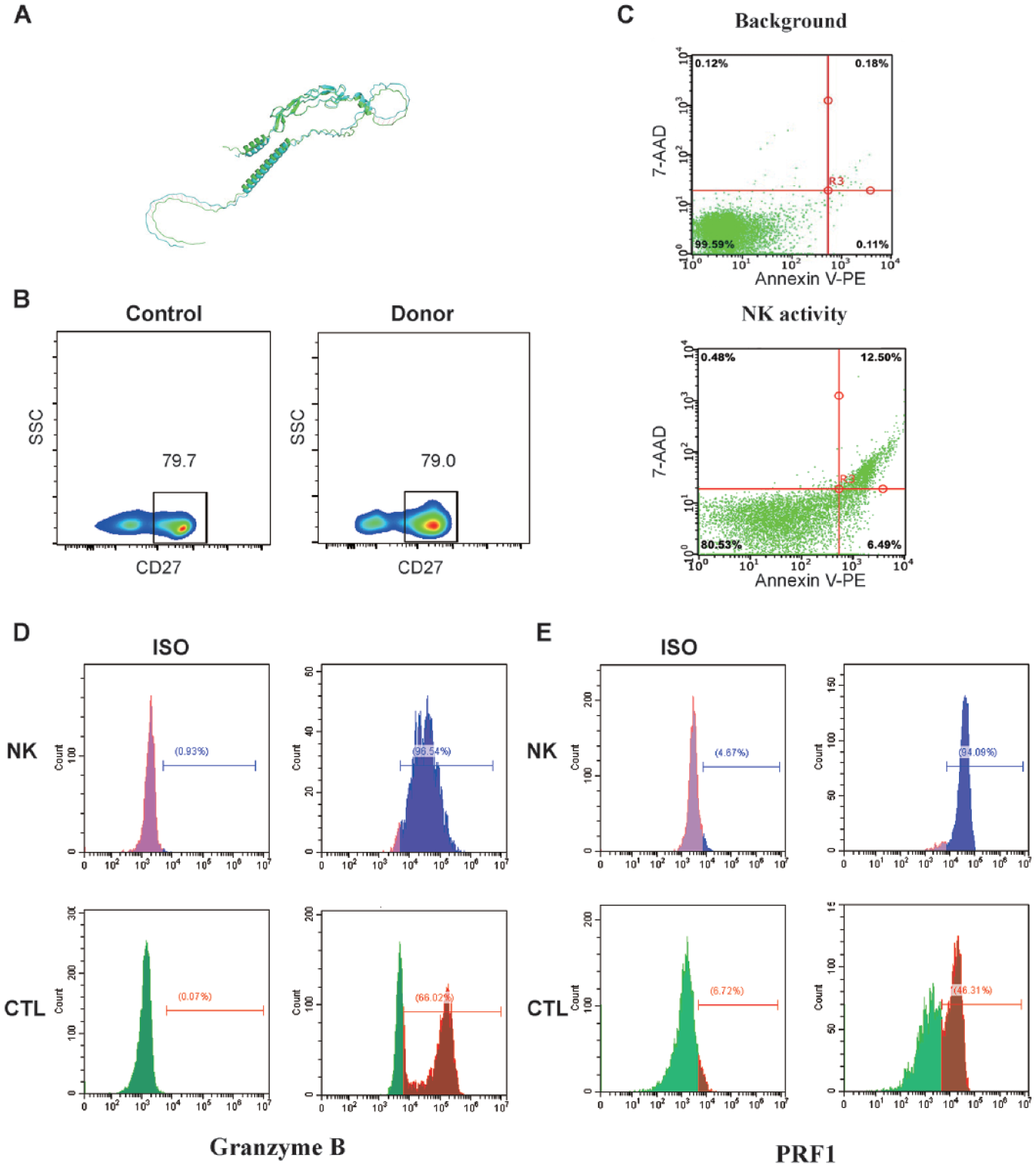
(A) Overlay of AlphaFold structures for WT (green model) and mutated CD27 (blue model). (B) CD27 surface expression on T cells from healthy controls (left panel) and donors (right panel). (C–E) Flow cytometry analysis of natural killer cell activity (C), granzyme B (D), and perforin (E) in patients with HLH. Left panels show the isotype control (ISO). CTL: cytotoxic T-lymphocytes; HLH: hemophagocytic lymphohistiocytosis; NK: natural killer; SSC: side scatter; WT, wild type; 7-AAD, 7-aminoactinomycin D.
In June 2022, he underwent allo-HSCT from his older brother with a myeloablative conditioning regimen consisting of cytarabine (2 g/m2/d, days −8 to −7), busulphan (0.8 mg/kg/q6h, days −6 to −4), cyclophosphamide (1.8 g/m2/d, days −3 to −2), and MeCCNU (250 mg/m2/d, day −1). A total of 6 × 108/kg mononuclear cells and 8.1 × 106/kg CD34+ cells were infused. Graft-versus-host disease (GVHD) prophylaxis included cyclosporine, mycophenolate mofetil, and short-term methotrexate. Neutrophil and platelet engraftment occurred on days +15 and +27, respectively. Chimeric analysis at 1, 2, 3, and 6 months post-transplant revealed persistent full-donor chimerism. PET/CT at 5 months after transplantation confirmed complete remission of disease (Fig. 1B). Three months post-transplantation, he developed grade II acute GVHD (aGVHD) manifesting with abdominal pain, diarrhea, and watery stools, which significantly alleviated after immunosuppressive agents treatment. At 10.9 months post-transplant, he was diagnosed with a COVID-19 infection, and his clinical condition improved after treatment. As of this report, no evidence of HLH or lymphoma was observed 15.2 months after HSCT.
Discussion
The pathogenicity of adult-onset HLH has been extensively explored in previous studies6,17–20. Zhang et al. 6 reported that 14% of adult-onset HLH cases exhibit hypomorphic heterozygous genetic variants (PRF1, MUNC13-4, or STXBP2). Sieni et al. 18 identified pathogenic variants of PRF1, UNC13D, STXBP2, and SH2D1A in 11 adult patients with familial hemophagocytic lymphohistiocytosis (FHL). Unlike pediatric cases where homozygous mutations or compound heterozygous variants are commonly detected, adult HLH cases typically involve heterozygous mutations, predisposing individuals to HLH after certain triggers 21 . In contrast to FHL, disruptive germline variants do not appear to be the primary drivers of adult HLH. In addition, there are no substantial clinical differences observed between patients with or without disruptive mutations 21 . However, the susceptibility of different defective genetic backgrounds to secondary factors remains obscure. In our study, we identified HLH-associated heterozygous missense mutations in UNC13D and CD27. Functional analyses consistently revealed impaired CD107a degranulation of NK and CTL but normal NK activity and perforin levels, indicating that his residual NK- and T-cell function may be sufficient to prevent the occurrence of clinical HLH for many years. The EBV infection and lymphoma development further triggered the pathogenesis and progression of HLH episodes. Other mutations in the patient, including those in NOTCH2 and NOTCH3, have also been implicated in the occurrence and progression of lymphomas, potentially synergizing deleterious effect with HLH-specific mutations 22 .
Consolidative allo-HSCT has emerged as the primary therapeutic approach for patients with pHLH and R/R sHLH. In a comprehensive retrospective study of 68 adult patients with HLH, patients undergoing allo-HSCT demonstrated a significantly improved median survival of 21.5 months 23 . Recent findings by Gooptu et al. presented encouraging outcomes for 21 adult HLH patients undergoing allogeneic HSCT, with 16 harboring one or more HLH-specific gene variants. The 3-year overall and progression-free survival rates stood at 75% and 71%, respectively, with no isolated HLH relapses recorded 24 . The critical role of donor screening for HLH-associated genes in subsequent HSCT was underscored in patients with defective gene expression. Genetic analysis of the patient’s family revealed inheritance of UNC13D and CD27 mutations from his parents. Notably, his brother shared the CD27 missense mutation without manifesting HLH symptoms. In the context of donor selection, the patient’s father, with UNC13D mutations, was deemed unsuitable, and the mother was excluded due to a higher prevalence of genetically susceptible genes and advanced age. Despite the older brother carrying some germline mutations, including the HLH-specific CD27 missense mutation, he emerged as a potential donor in the absence of alternative options. CD27 is a member of the tumor necrosis factor receptor family and is expressed on a broad range of human lymphocytes, including T cells, germinal centers, memory B cells, and some NK cells 25 . In addition, CD27 provides co-stimulatory signals that potentiate T-cell activation, survival, proliferation, and differentiation upon binding to its specific ligand, CD70. Although the literature on CD27 variants is limited, our study identified a novel heterozygous CD27 variant. Through meticulous examination of T-cell function via flow cytometry and functional prediction, we determined this variant to be of uncertain significance, with no discernible impact on T- or NK-cell functions. Consequently, the patient successfully underwent HSCT from his brother and remained free of lymphoma and quiescent HLH.
Conclusion
In conclusion, a thorough understanding of both the functional and genetic facets of HLH genetic defects is essential, which provides a rationale for implementing appropriate therapeutic interventions and the judicious selection of transplant donors. Our case underscores the significance of early allogeneic hematopoietic stem cell transplantation (allo-HSCT) as a curative strategy for germline gene mutations, contributing to sustained long-term remission.
Supplemental Material
sj-docx-1-cll-10.1177_09636897231221887 – Supplemental material for Relapsed/Refractory Peripheral T-Cell Lymphoma-Associated Hemophagocytic Lymphohistiocytosis With UNC13D and CD27 Germline Mutations
Supplemental material, sj-docx-1-cll-10.1177_09636897231221887 for Relapsed/Refractory Peripheral T-Cell Lymphoma-Associated Hemophagocytic Lymphohistiocytosis With UNC13D and CD27 Germline Mutations by Tingting Yang, Rongrong Chen, Mingming Zhang, Ruirui Jing, Jia Geng, Guoqing Wei, Yi Luo, Pingnan Xiao, Ruimin Hong, Jingjing Feng, Shan Fu, Houli Zhao, Jiazhen Cui, Simao Huang, He Huang and Yongxian Hu in Cell Transplantation
Footnotes
Acknowledgements
We thank the patients and their parents for participating in this study.
Author Contributions
TY and RC collected data and drafted the manuscript. MZ, RJ, GW, YL, PX, RH, JF, SF, and HZ provided patient care and helped collect and discuss the clinical data. JC and SH performed the functional analyses. JG provided Imaging data. HH and YH supervised and added important intellectual content. All authors have reviewed and approved the final draft of the manuscript.
Ethical Approval
This study was approved by the Institutional Review Board of the First Affiliated Hospital, School of Medicine, Zhejiang University in accordance with the Declaration of Helsinki (as revised in 2013). The manuscript was not presented at a podium or poster meeting presentation.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
The patient and family gave informed consent to participate in the study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by grants from the National Natural Science Foundation of China (82270234 and 82130003) and Key Project of Science and Technology Department of Zhejiang Province (2021C03010 and 2023C0306).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.