
Abstract
Cooling at 4°C is routinely used to lower metabolism and preserve cell and tissue integrity in laboratory and clinical settings, including organ transplantation. However, cooling and rewarming produce cell damage, attributed primarily to a burst of reactive oxygen species (ROS) upon rewarming. While DNA represents a highly vulnerable target of ROS, it is unknown whether cooling and/or rewarming produces DNA damage. Here, we show that cooling alone suffices to produce extensive DNA damage in cultured primary cells and cell lines, including double-strand breaks (DSBs), as shown by comet assay and pulsed-field gel electrophoresis. Cooling-induced DSB formation is time- and temperature-dependent and coincides with an excess production of ROS, rather than a decrease in ATP levels. Immunohistochemistry confirmed that DNA damage activates the DNA damage response marked by the formation of nuclear foci of proteins involved in DSB repair, γ-H2Ax, and 53BP1. Subsequent rewarming for 24 h fails to recover ATP levels and only marginally lowers DSB amounts and nuclear foci. Precluding ROS formation by dopamine and the hydroxychromanol, Sul-121, dose-dependently reduces DSBs. Finally, a standard clinical kidney transplant procedure, using cold static storage in UW preservation solution up to 24 h in porcine kidney, lowered ATP, increased ROS, and produced increasing amounts of DSBs with recruitment of 53BP1. Given that DNA repair is erroneous by nature, cooling-inflicted DNA damage may affect cell survival, proliferation, and genomic stability, significantly impacting cellular and organ function, with relevance in stem cell and transplantation procedures.
Introduction
Cooling is a frequently used procedure to lower metabolism and preserve cells, both in the laboratory setting and in the clinics, for instance, to limit organ damage in transplantation, major surgery, and following infarction1–3. In terms of cellular stress, cooling–rewarming resembles ischemia-reperfusion (I/R) injury, during which cells initially suffer from impaired nutrient and oxygen supply, and followed by the rapid generation of reactive oxygen species (ROS) during reperfusion4–6. Similarly, cooling and rewarming produce cell damage and cell death5,7. Consequently, similar to I/R, most mammalian cell types are vulnerable to prolonged and profound cooling attributed primarily to a burst of ROS upon rewarming5,8,9. However, we recently showed oxidative damage to occur during the cooling phase, as lowering temperature results in a continued production of ROS, a concomitant failure of endogenous antioxidant capacity, and lipid peroxidation10,11. The latter observation implies that cooling alone may suffice to confer oxidative damage to various macromolecules.
Excessive formation of ROS, specifically the hydroxyl radical (•OH), is also known to damage DNA resulting from the abstraction of a deoxyribose hydrogen atom from its sugar-phosphate backbone12,13. DNA represents a highly vulnerable target of ROS as it is the only biomolecule that cannot be replaced by de novo synthesis. Cooling and rewarming were previously reported to induce chromatin condensation and DNA fragmentation after rewarming, which likely reflect ROS-mediated induction of apoptosis 7 . In addition, chromatin condensation may result from chromatin-modifying enzyme activity because of enzyme inhibition, or indirectly via energy depletion or interference with nuclear transport. However, ROS are also expected to induce direct single-strand breaks (SSBs) and oxidative DNA lesions which may result in double-strand breaks (DSB). In addition, ROS may induce direct DNA damage in the form of SSB and DSB following damage to the DNA backbone 14 or oxidation of DNA-associated proteins 15 . The ensuing DNA damage response (DDR), consisting of homologous recombination (HR) and non-homologous end-joining (NHEJ), the latter displaying a high incidence of errors, may ultimately lead to genome instability16–18. Generally, ROS-induced DNA damage contributes to carcinogenesis, aging, and neurodegeneration (see, for review, the work by Niedernhofer et al. 19 ). Moreover, oxidative modification of guanine in promotor regions may substantially increase gene expression 20 . In addition to ROS-induced DNA damage, ATP depletion during cooling and rewarming may further promote activation of the DDR and apoptosis as demonstrated in I/R injury8,21,22.
Cooling and rewarming typically induce extensive cell death in cells from nonhibernators, such in contrast to hibernator cells, which have escaped vulnerability to cooling because of cell-autonomous mechanism(s) 9 , likely involving a limitation of mitochondrial ROS production11,23,24. One of the mechanisms conferring resistance to cooling is preservation of endogenous H2S production by reuptake of biogenic amines excreted during cooling 9 . Dopamine9,25,26 and hydroxychromanol derivatives, including Sul-121 and Sul-10927,28, have similar effects both in vitro and in vivo, thus fully abrogating oxidative stress–induced cell death during cooling with or without subsequent rewarming. Whether these compounds also counteract cooling and rewarming–induced DNA damage is unknown.
Here, we explore cooling and rewarming effects on DNA strand breaks in various cultured primary cells and cell lines, and in a renal transplantation model using static cold storage of porcine kidney. We show that cooling of cells and kidney precipitates both SSBs and DSBs in a time- and temperature-dependent fashion, related to increased formation of ROS. Moreover, in cells we observed a lack of DNA repair during subsequent rewarming, despite activation of the DDR, which was associated with an absence in recovery of ATP synthesis. Finally, we show that limiting ROS production by dopamine and Sul-121 concentration dependently protects from cooling-evoked DNA damage and cell death.
Materials and Methods
Cell Culture, Cooling and Rewarming Induction
Rat vascular smooth muscle cells (A7R5, ATCC CRL1444, USA) and human liver cancer cell (HEPG2 kind gift of Dr. H. Moshage) were cultured at 37°C in 5% CO2 in air in DMEM (Dulbecco’s Modified Eagle Medium BRL 41966-029; Gibco, the Netherlands) and rat smooth muscle aortic cells (SMAC; ATCC CRL1476) in DMEM/F12 (Gibco), all supplemented with 10% (v/v %) heat-inactivated fetal calf serum (FCS) and 100 U ml−1 penicillin. Human umbilical vein endothelial cells (HUVEC) were obtained from the Endothelial Cell Facility of the UMCG (University Medical Center Groningen) and cultured in the supplied EC medium containing 20% (v/v %) heat-inactivated FCS supplemented with penicillin/streptomycin. Cells were plated in six-well plates and grown to 80% confluency. Standard cooling and rewarming (C/R) protocol consisted of 24 h cooling at 4°C by placing cells in a fridge, resulting in gradual reduction of medium temperature in about 40 min (~25 min to 10°C), followed by 4 h of rewarming. Influence of cooling temperature was additionally examined in cells exposed to 8, 16, and 24°C for 24 h, while the influence of duration of cooling was examined at 4, 8, 16, and 24 h in cells cooled at 4°C. The effect of different duration of normothermia after rewarming was assessed at 1, 4, and 24 h post-rewarming. The effect of drugs on cell viability was examined in SMAC treated from 30 min prior to cooling with dopamine (0.3, 3, 30 µM) and Sul-121 (0.001, 0.01, 0.1 µM) and throughout C/R. Cell viability was assessed by trypan blue and neutral red (NR) assays. To quantify dead and alive cells, they were incubated in a 0.4% solution of trypan blue in phosphate-buffered saline (PBS) of pH 7.2 (Sigma-Aldrich, Amsterdam, the Netherlands) and blue and total cells were counted in a Bürker chamber. NR assay was performed following replacement of normal media by NR media [culture media with 5% FBS (fetal bovine serum) and 50 mg/ml NR dye; Sigma-Aldrich]. Next, cells were lysed and absorbance was measured at 450 nm using a Synergy 2 Multi-Mode plate reader (BioTek, Landsmeer, the Netherlands).
Porcine Kidney Model
Dutch Landrace pig (90–110 kg) kidneys were harvested and a biopsy was taken. Subsequently, they were flushed with ice-cold preservation solution of the University of Wisconsin (UW) 2 . Thereafter, kidneys were placed in a plastic bag containing UW solution, which was placed in a polystyrene box on ice. Further biopsies were taken from the cold kidney after 8, 16, and 24 h of cooling. This procedure was reported to result in similar cooling rates as observed in cells, ie, 10 to 18 min to 10°C 29 .
Comet Assay
Cells were obtained after trypsinization and loaded on gel following lysis according to the manufacturer’s instructions (Trevigen CometAssay Kit, 4250-050-K; Trevigen). Alkaline comet assay, detecting both single DNA strand breaks and double DNA strand (SSB + DSB), and neutral comet assay, detecting DSB, were performed to quantify and classify DNA damage according to the manufacturer’s instructions. Per condition, >100 comets were photographed (40× magnification, Leica DM2000 LED (Wetzlar, Germany)) and quantified by expressing DNA damage as the percentage of DNA in the comet’s tail (%tailDNA) using ImageJ software (Bagnell, R. Comet Assay ImageJ Macro. http://www.med.unc.edu/microscopy/resources/imagej-plugins-and-macros/comet-assay) as exemplified in Fig. S1.
Pulsed-Field Gel Electrophoresis
Electrophoresis was performed using a CHEF DR II-apparatus (Biorad, Lunteren, the Netherlands) with a hexagonal array of 24 electrodes producing a field reorientation angle of 120° 4 . Plugs made from 2% (v/v %) agarose (chromosomal grade agarose; Biorad) containing cells (~ 1.5–2 × 107 cells/ml), lysed [0.5 M EDTA, 1% Sarcosyl (pH 9), 0.5 mg ml−1 proteinase K for 2 days at 50°C] and inserted into 0.8% gel agarose (chromosomal grade agarose; Biorad) in Tris-borate-EDTA (TBE; pH 8). Gels ran at 120° in TBE at 14°C for 24 h using a linear pulse time of 75 min and a field strength of 1.2 V/cm.
Immunofluorescent Staining
Cells were cultured on glass coverslips, washed with PBS, and then fixed with 3% paraformaldehyde in PBS for 10 min. Fixed cells or tissue sections were rinsed with PBS and permeabilized with 0.1% Triton-X-100 for 10 min, washed and incubated for 1 h with PBS/1% BSA (bovine serum albumin) following which cells were incubated 1 h at room temperature or overnight at 4°C with anti-53BP1 polyclonal antibody (diluted 1:500, H-300, Santa Cruz (Heidelberg, Germany)) and/or anti-γ-H2Ax polyclonal antibody (phospho S139, Abcam, diluted 1:100). After rinsing with PBS/0.05% Tween 20, coverslips were incubated with secondary antibodies (Southern Biotech, IgG-FITC, and LifeTechnologiesTM Alexa Fluor, TRITC, both diluted 1:500) in PBS/5% BSA for 1 h. After a PBS/0.05% Tween 20 wash, DNA was counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich) for 10 min and coverslips were mounted in Fluorescent Mounting Medium (Dako) and imaged using a standard fluorescence microscope (Leica DM2000 LED). Specificity of staining was verified using untreated and γ-radiation–exposed cells and tissue (4 Gray). Foci counts were assessed manually and solely represent colocalized signals of 53BP1 and γ-H2Ax and were analyzed in 2D; signals that did not co-localize were ignored, irrespective of size or intensity. Per condition, >100 nuclei were photographed and the number foci/nucleus quantified.
ATP and Lipid Peroxidation
Cells or tissue were harvested by addition of EDTA buffer, cell scraping on ice and boiling for 6 min. ATP was measured with a luciferase assay (Promega, Leiden, the Netherlands) with luminescence measured at 590 nm. Lipid oxidation was quantified by measurement of malondialdehyde (MDA) using the OxiSelect TBARS assay kit (Cell Biolabs, San Diego, USA). ATP and lipid peroxidation levels were corrected for protein levels (Bradford assay; Biorad).
Statistical Analysis
Data are represented as mean ± SEM, unless indicated otherwise. Statistical data analyses were performed using one-way ANOVA (analysis of variance) with Tukey’s test with P < 0.05 considered statistically significant (GraphPad Prism 7). Differences between comet assay distributions were tested using the R package nparcom 30 using R version 3.6.2 31 .
Results
Cooling Confers Time- and Temperature-Dependent DNA Strand Breaks
To examine the effect of cooling and rewarming on DNA strand breaks in different cell lines, we performed alkaline single-cell gel electrophoresis (comet assay) and quantified the percentage of DNA in the tail of comets (%tailDNA) in primary human endothelium cells (HUVEC), human hepatocellular carcinoma cell line (HEPG2), rat smooth muscle cell line (A7r5), and rat smooth muscle primary cells (SMAC) cooled at 4°C for 24 h with and without rewarming for 4 h. Cooling induced a large increase in median %tailDNA, increasing from baseline levels of 3% to 15% to 53% to 83% (Fig. 1A). Subsequent rewarming for 4 h resulted in a further increase of median %tailDNA in HUVEC to 89%, and a reduction in HEPG2 (48%), A7r5 (30%), and SMAC (67%). To further examine cooling and rewarming–induced DNA damage, comet assays were obtained in SMAC both under alkaline and neutral conditions, representing the total of single- and double-strand breaks (SSB+DSB) and solely DSB, respectively. Cooling (24 h, 4°C) of SMAC increased the median %tailDNA from 6% at baseline to 78% in alkaline comets, which reduced to 67% following rewarming (Fig. 1B, D). Similarly, in neutral comets, median %tailDNA increased from 7% at baseline to 56% following cooling, with a subsequent modest reduction to 49% following rewarming (Fig. 1B, D). To confirm that cooling induced DSB, pulsed-field gel electrophoresis (PFGE) was performed. In agreement with results of the neutral comet assay, cooling and rewarming substantially increased DNA smearing compared with noncooled cells (Fig. 1C, E). It is of note that cooling reduced cell viability by 15% and 21%, as measured, respectively, by NR assay and trypan blue staining (Supplemental Fig. S2). However, even if this cell loss represented solely undamaged cells, the resulting ~25% enrichment in remaining cells cannot explain the much larger increase in cells with DNA strand breaks. In agreement, cooling strongly lowered caspase 3/7 activity compared with control and rewarmed SMAC, to a level which was unaffected by the pan-caspase inhibitor Z-VAD-FMK (50 µM, Supplemental Fig. S3). Together, these results demonstrate that cooling induces substantial amounts of DNA strand breaks largely consisting of DSB, which mostly persist after rewarming.

Effect of cooling and rewarming on DNA strand breakage.(A) DNA strand breaks assessed with alkaline comet assay in HUVEC, HEPG2, A7r5, and SMAC cells. DNA strand breaks were quantified in individual cells as % DNA in the comet tail over total DNA (%tailDNA) in noncooled control cells (37°C), cells cooled for 24 h at 4°C (C), and cooled cells following an additional rewarming for 4 h at 37°C (C/R4). Median %tailDNA is indicated by horizontal line. (B) Typical examples of comets at 40× magnification. (C) Typical example of pulsed-field gel electrophoresis lanes in noncooled control SMAC (37°C) and 24 h cooled cells followed by rewarming for 4 h at 37°C (C/R4). (D) Quantification of DNA strand break under alkaline (SSB+DSB) and neutral (DSB) conditions in SMAC. Median %tailDNA is indicated by red horizontal line. (E) Quantification of DNA smear. Results are given as mean ± SEM. Comets were quantified in >100 cells. C/R: cooling and rewarming; DSB: double-strand breaks; SSB: single-strand breaks. ***P < 0.001; *different from 37°C; #different from C/R4, *P < 0.01.
Cooling-Induced DNA Strand Breaks Are Time- and Temperature-Dependent and Relate to Free Radical Production
Next, to determine the influence of different cooling temperatures on DNA damage, SMAC were incubated for 24 h at temperatures ranging from 37°C to 4°C (Fig. 2A). Cooling from 37°C down to 8°C only marginally increased %tailDNA from 8% to 28% in both alkaline and neutral comet assays (Fig. 2A). In contrast, cooling to 4°C induced a sharp increase in %tailDNA in alkaline comet assay (77%) with a smaller increase under neutral conditions (53%). Thus, cooling at 4°C resulted in an increase in both SSB and DSB, whereas moderate cooling induced mainly DSB, perhaps resulting from still ongoing repair of SSB. Next, the time course of induction of DNA strand breaks was examined in SMAC by obtaining comet assays at 4, 8, 16, and 24 h of cooling at 4°C. Whereas median %tailDNA at 4 and 8 h was similar to baseline, longer cooling up to 24 h resulted in a gradual increase in %tailDNA both in alkaline and neutral comets (Fig. 2B). To examine DNA repair after rewarming in more detail, cells were cooled for 24 h at 4°C with increasing duration of rewarming up to 24 h. Compared with cooled cells, median %tailDNA of alkaline comets decreased substantially at 1 and 24 h of rewarming, but increased in between at 4 h (Fig. 2C). In contrast, median %tailDNA of neutral comets showed a similar decrease at all time points after rewarming. Collectively, these data show that cooling at lower temperature and during longer time periods increases DNA damage, with persistence of both SSB and DSB during prolonged rewarming.
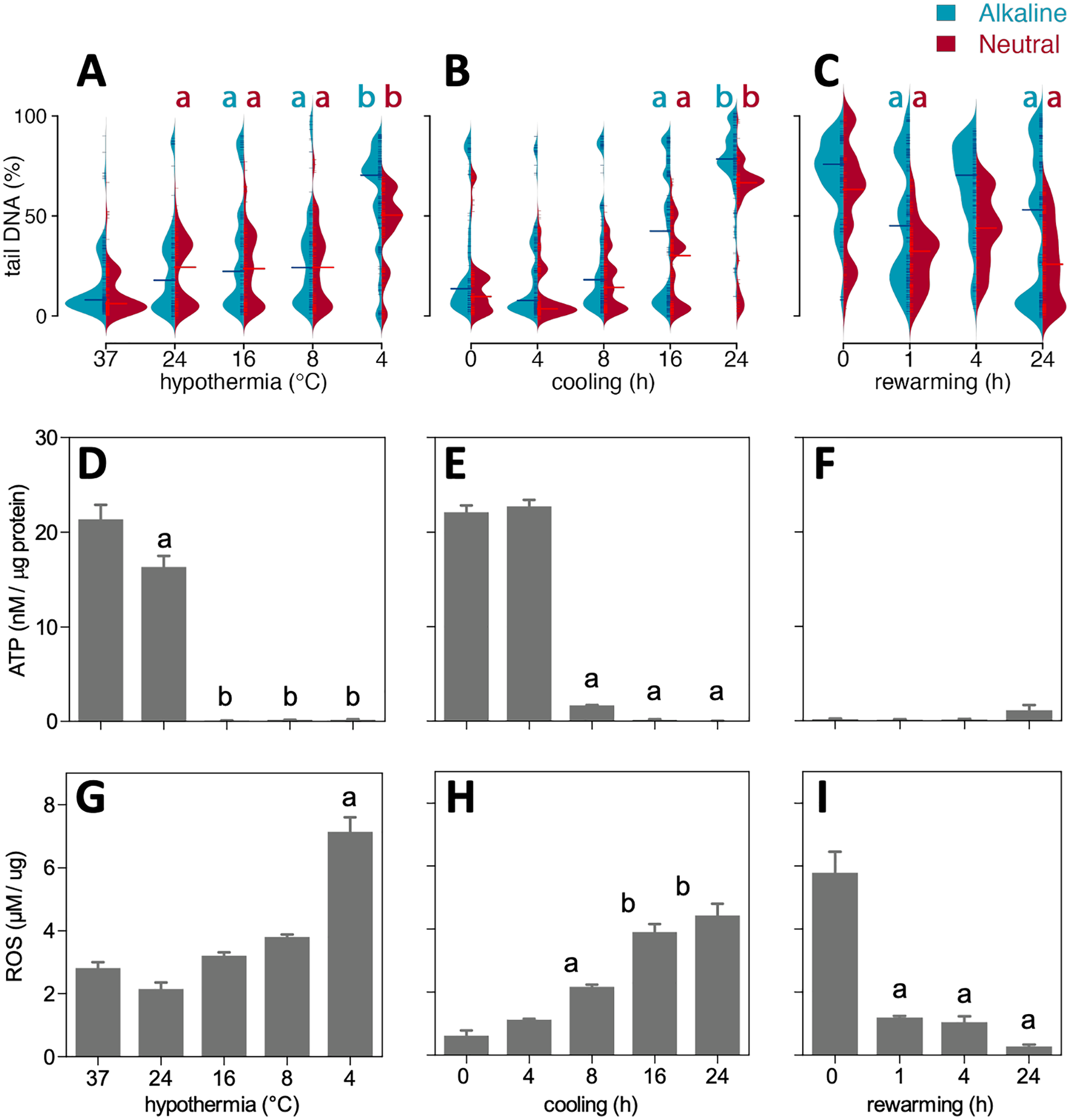
Time and temperature dependency of DNA stand breaks in cooled and rewarmed SMAC. DNA strand breaks were quantified in individual cells as % DNA in the comet tail over total DNA (%tailDNA). (A) 24 h cooling at different temperatures (37, 24, 16, 8, and 4°C). (B) Cooling at 4°C during 0, 4, 8, 16, and 24 h. (C) 24 h of cooling at 4°C followed by rewarming for 0, 1, 4, and 24 h. (D–F) Cell ATP concentrations corrected for protein levels in conditions indicated in (A)–(C). (G–I) ROS levels measured as lipid peroxidation corrected for protein levels in conditions indicated in (A)–(C). Data are mean ± SD; a, b denote statistical differences between groups. ROS: reactive oxygen species.
We exploited the variation in DNA strand breaks in different conditions to examine potential cause(s) of cooling-induced DNA damage and examined concomitant changes in energy reserves and free radical production by quantifying ATP and ROS levels in SMAC. Cooling for 24 h at 24°C modestly reduced ATP levels, whereas ATP dropped below the detection limit after cooling at 16°C and below (Fig. 2D). In contrast, ROS production was only increased after cooling at 4°C (Fig. 2G). Cooling at 4°C for different time periods resulted in a large drop in ATP levels at 8 h and longer (Fig. 2E), whereas ROS levels showed a gradual linear increase over time (Fig. 2H). Next, we tested whether ROS and ATP normalized to baseline after rewarming for up to 24 h. Interestingly, ATP levels remained low up to 24 h of rewarming (Fig. 2F), whereas ROS levels normalized after 1 h of rewarming and beyond (Fig. 2I). These data suggest that DNA strand breaks are caused by excess ROS production during cooling, while incomplete repair of DSB during rewarming is mainly caused by nonrecovery of ATP production.
Dopamine and Sul-121 Dose-Dependently Attenuate Cooling-Induced DNA Strand Breaks
We and others previously showed that incubation with dopamine or 6-hydroxychromanols precludes cooling-induced cell death in various cell lines9,27,32 and in in vivo deep cooling of rat26,28. Therefore, we next examined the reduction of DNA damage by dopamine (0.3–30 µM) and Sul-121 (1–100 nM) in SMAC cooled for 24 h at 4°C and treated from 30 min prior to cooling onward (Fig. 3A, B). Both dopamine and Sul-121 concentration-dependently attenuated DNA damage, as reflected in a gradual decrease of %tailDNA in both alkaline and neutral comets. Notably, both compounds were more potent in precluding SSB than DSB and did not promote DSB repair during 4 h of rewarming (Fig. 3C), corroborating their antioxidant effect as their mechanism of action. The lowering of the amount of DSBs by dopamine and Sul-121 assessed by comets was corroborated by PFGE, showing both compounds to reduce DNA smearing in SMAC (Fig. 3D, E).

Dopamine and Sul-121 concentration-dependently prevent DNA strand breaks in cooled and rewarmed SMAC. (A, B) Typical examples of alkaline and neutral comets at 40× magnification. Noncooled control SMAC (37°C), SMAC cooled for 24 h at 4°C (C), and cooled SMAC with an additional rewarming for 4 h at 37°C (C/R4) were examined in the presence of vehicle, dopamine (Dopa) and Sul-121. (C) Quantification of DNA strand breaks under alkaline (SSB+DSB) and neutral (DSB) conditions in noncooled control cells (37°C) and cells cooled for 24 h at 4°C (“Cooled”) and cooled cells following an additional rewarming for 4 h at 37°C (“Rewarmed”) in absence and presence of dopamine and Sul-121. Dose response curves are mean and range (25th and 75th percentile). (D, E) Pulsed-field gel electrophoresis of C/R4 SMAC in the absence or presence of dopamine (Dopa; 30 μM) or Sul-121 (100 nM). a, b, c denote differences from other groups. C/R: cooling and rewarming; DSB: double-strand break; SSB: single-strand break.
To substantiate that cooling and rewarming induce a DDR, the accumulation of nuclear DNA damage foci that were double positive for γ-H2Ax and 53BP1 was quantified in SMAC cooled at 4°C for 24 h and rewarmed for 1, 4, and 24 h, both in the absence and presence of dopamine and Sul-121. Cooling sharply increased the number of double-stained foci/nucleus from 1.5 at baseline to 11.8 following cooling and 13.7 following 1 h rewarming, with further rewarming for 4 and 24 h only marginally reducing the number of double-stained foci/nucleus (Fig. 4A–C, Supplemental Fig. S3). Both treatment with dopamine and Sul-121 significantly reduced double-stained foci accumulation after 24 h cooling and following rewarming up to 24 h of SMAC (Fig. 4D, E, Supplemental Fig. S4). Thus, accumulation of 53BP1 and γ-H2Ax corroborates cooling-induced DNA damage, absence of substantial DNA repair during rewarming, and the protective effect of dopamine and Sul-121.
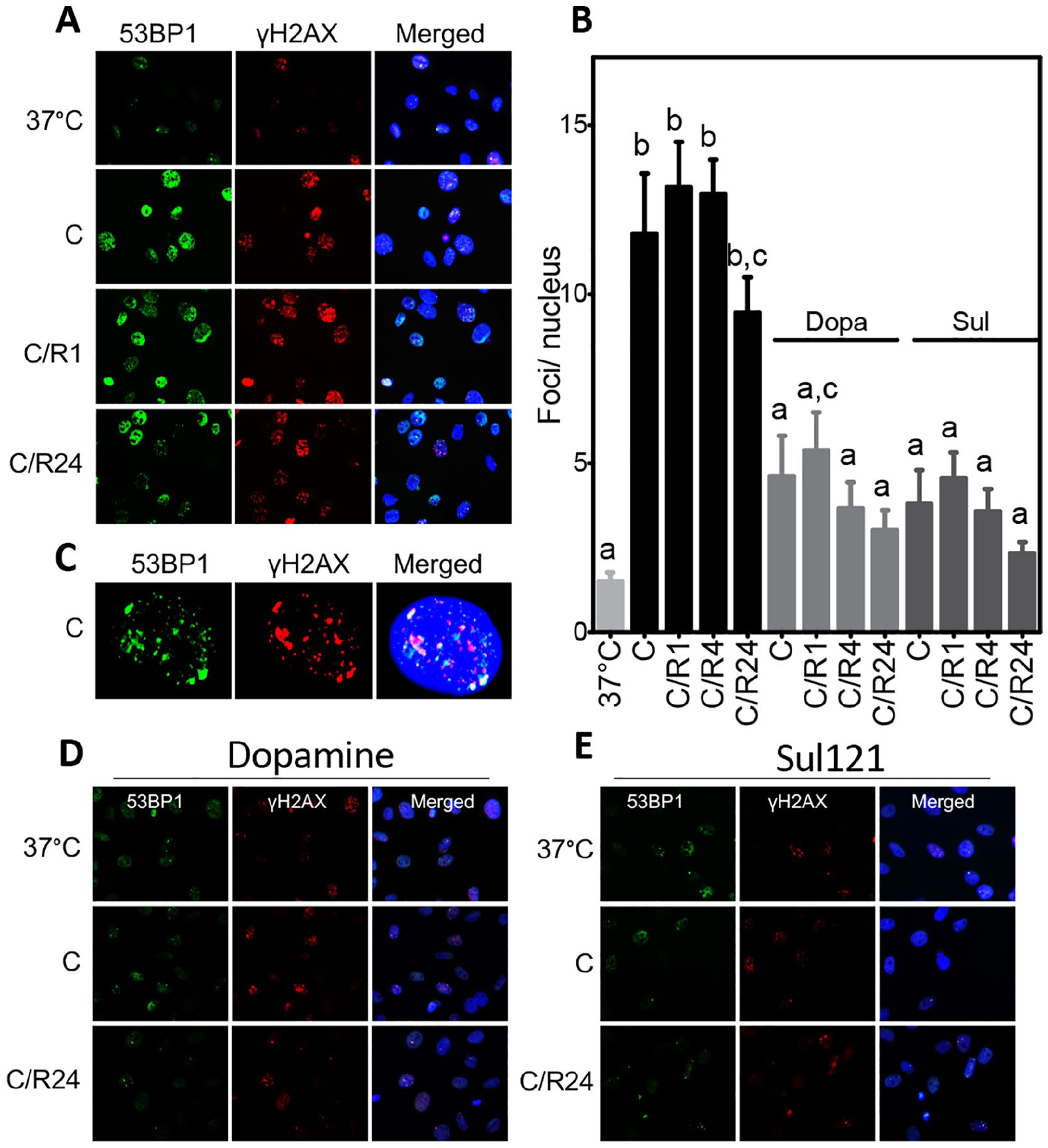
Immunofluorescent staining of γH2AX and 53BP1. (A) Nuclear foci of γH2AX (red) and 53BP1 (green) increase after 24 h of cooling (C) at 4°C and persist during subsequent rewarming for 1, 4, and 24 h (C/R1, 4, 24) of SMAC. Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI; blue). (B) Quantification of double-stained foci/nucleus. (C) Typical example of single nucleus (blue) of cooled SMAC, showing co-localization of γH2AX and 53BP1. (D, E) 53BP1 and γH2AX staining of SMAC treated with dopamine (Dopa) or Sul-121. a–c denote differences from other groups. C/R: cooling and rewarming.
DNA Damage During Cold Static Kidney Transplant Preservation
Clinically, deep hypothermia is used in organ transplantation, with static cooling at 4°C routinely applied to preserve the organ after procurement and during transport 1 . To explore effects of cooling on DNA damage in this setting, a standard transplantation procedure of cold storage in UW solution 33 on ice was used to preserve freshly procured porcine kidney up to 24 h. Tissue DNA damage was examined by PFGE and immunohistochemistry for 53BP1, in addition to measurement of ROS (MDA assay) and ATP levels. Similar to cells, PFGE of 4°C stored kidney shows DNA smearing to increase after 8 h of cooling (Fig. 5A, B), which was paralleled by a decrease in ATP levels (Fig. 5C) and an increase in ROS (Fig. 5D). Furthermore, the number of nuclear foci of 53BP1 was dramatically increased by 24 h cold storage (Fig. 5E, F). Similar to cells, extended cooling of kidney under standard transplantation conditions induced double-strand DNA breaks, ATP depletion, ROS accumulation, and activated DNA repair.
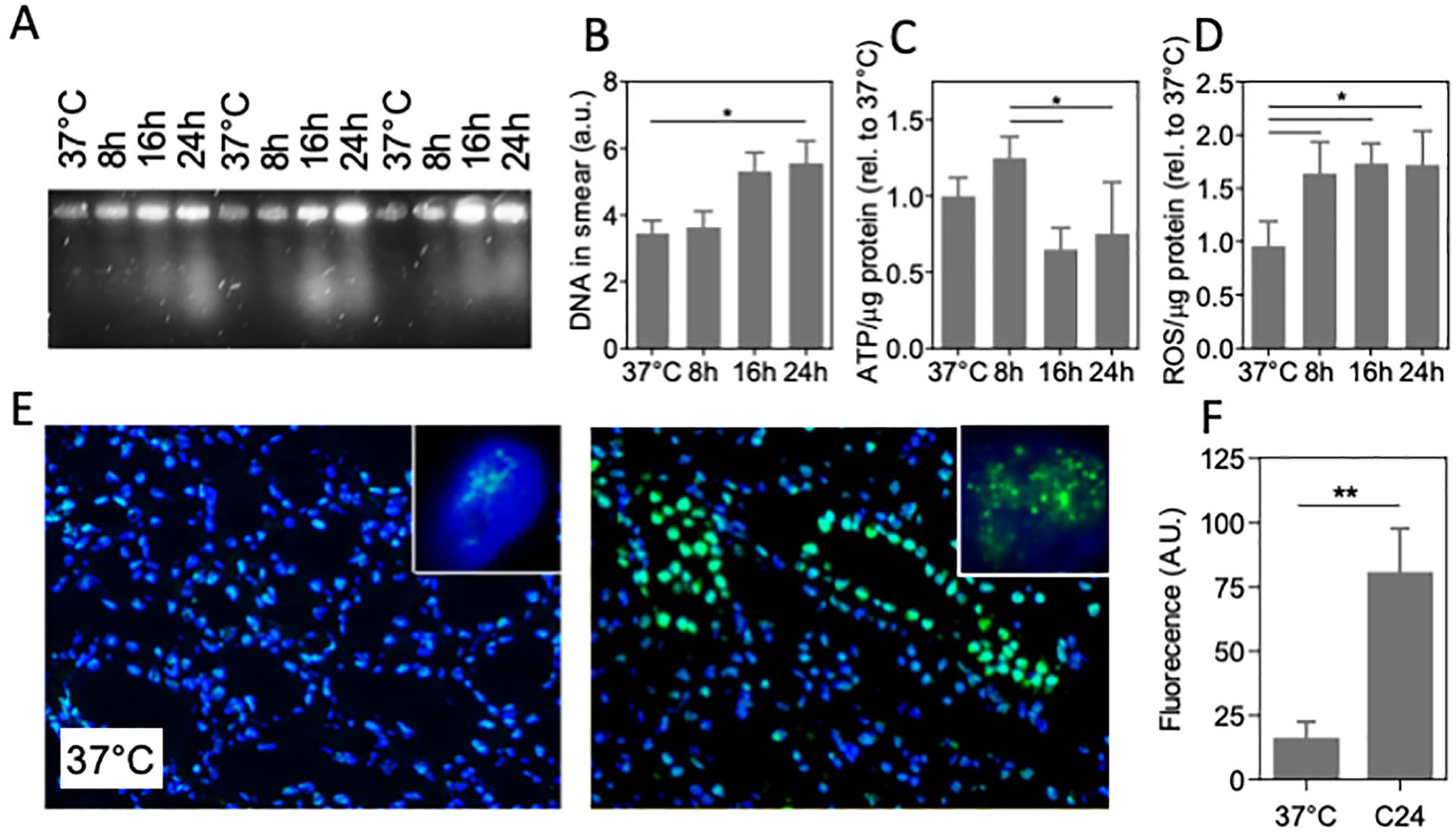
DNA damage in cold-preserved porcine kidney. (A) Representative pulsed-field electrophoresis performed on pig kidney at 37°C and cooled kidney for 8, 16, and 24 h (4°C, UW preservation solution; three different kidneys shown). (B) Quantification of the amount of DNA in the PFGE smear. (C) ATP levels normalized to protein abundance. (D) Quantification of ROS levels assessed by MDA assay normalized to protein abundance. (E) Typical example of fluorescent staining of immunofluorescent staining of 53BP1 staining (green) of pig kidney tissue, showing abundant nuclear foci after 24 h cooling at 4°C. Nuclei are stained with 4′,6-diamidino-2-phenylindole (DAPI;blue). Insets: representative single nucleus of a tubular cell. Original magnification: 100×. (F) Quantification of fluorescent intensity of 53BP1 staining. MDA: malondialdehyde; PFGE: pulsed-field gel electrophoresis; ROS: reactive oxygen species; UW: University of Wisconsin. C24 denotes cooling for 24 h; *P < 0.05; **P < 0.01.
Discussion
Our results document extensive DNA damage during cooling in various cultured cells and in cold-stored kidney, without the need for rewarming. DNA strand breaks were evidenced both by comet assay and PFGE, and by an increase of DNA repair, ie, γ-H2Ax and 53BP1. Cooling-induced DNA strand breakage increased at lower cooling temperatures and with longer duration of cooling, in parallel with excess production of ROS. Increased levels of ROS during cooling originate from excess production due to a relative stronger inhibition of the mitochondrial electron transfer chain at its distal site than its proximal site, leading to escaping oxygen radicals10,11,23. Moreover, cooling impairs cellular antioxidant capacity, promoting to the accumulation of ROS in a time-dependent manner 10 . Furthermore, involvement of excess ROS levels is corroborated by the attenuation of cooling-induced DNA damage by dopamine and Sul-121. In addition, despite recruitment and activation of DNA repair factors, rewarming did not result in substantial repair of DNA breaks, likely due to persistent low ATP levels throughout 24 h of rewarming. Similar to cells, the standard static cold preservation procedure used during kidney transplantation provoked substantial DNA strand breakage and recruitment of repair factors in porcine kidney, with excess ROS production and ATP depletion. These results signify that avoiding deep and prolonged cooling limits DNA strand breaks, which will ultimately promote cell survival and maintain genomic integrity, both in the laboratory setting and in transplantation. Furthermore, we validated compounds capable of precluding DNA damage, should deep or longer cooling be required.
While previous studies showed cooling to induce apoptosis and the related blebbing, chromatin condensation, and DNA fragmentation in hepatocytes and hepatic endothelial cells following their rewarming 7 , our study is the first to document that DNA strand breaks are precipitated during cooling, without the need of rewarming. Cooling thus provokes both SSBs and DSBs, with an increasing number of strand breaks during deeper and more prolonged cooling. DNA strand breaks during cooling are unlikely to result from apoptosis, given the very low caspase activity and lack of effect of caspase inhibition we observed in cooled cells, and the absence of ATP, required in a number of apoptotic steps 34 . Moreover, the increase in SSBs and DSBs aligns with the increase in ROS production and not with the loss of ATP, implying that cooling-induced DNA damage results from increased DNA damage by ROS 17 rather than from deficiency of DNA repair because of low ATP levels. Given that ROS increases in cooled cells that are at low metabolic rate and that lipid peroxidation and apoptosis in cooled cells are counteracted by iron chelators7,25, the most likely origin of ROS is through production of •OH radicals by Haber-Weiss and/or Fenton reactions 12 . ROS insult is recognized as the main source of DNA lesions4,13,35. The critical role of ROS formation in cold-induced DNA strand breakage is corroborated by the protection conferred by dopamine and SUL-121, respectively, acting as an ROS scavenger 36 and inducer of H2S formation 9 , and by suppressing ROS formation through preservation of mitochondrial electron transport 27 . Furthermore, the gradual increase in ROS levels during prolonged cooling at 4°C in SMAC corroborates previous data documenting a similar phenomenon in hepatocytes, endothelial cells, and adipose-derived stem cells7,25,27. Interestingly, the rapid loss of ATP and slower increase in ROS in SMAC are consistent with previous findings in a pig kidney transplantation model employing cold machine perfusion, showing a strong reduction in the organ’s O2 consumption with only a moderate lowering of ROS levels at 4°C 10 .
Our results demonstrate that cooling suffices to induce a substantial amount of DSBs. This is evidenced primarily by the neutral comet assay and PFGE, both assessing the physical status of DNA. In addition, DSBs are corroborated by the formation of γ-H2Ax and 53BP1 foci, because of their specificity for DSB37–39. Oxidative damage can indeed produce DSBs, which would need relative high •OH concentrations40,41. In keeping, the sharp increase in DSB produced by cooling at 4°C and at 16 and 24 h cooling duration coincides with the major increases in ROS levels. DSB may also be secondary to SSBs and oxidative DNA damage during S-phase as a consequence of replicating the damaged DNA 42 . Alternatively, misregulation of transcription and RNA processing may lead to DSB formation via R loop formation as well43,44, possibly explaining the levels of DSB under more moderate cooling or at shorter cooling duration. Interestingly, similar patterns of co-localization of γ-H2Ax and 53BP1 foci were found previously in cryopreserved cells after thawing and shown to originate almost exclusively from collapsed replication forks of replicating (S-phase) cells 42 . Although solely cooled cells (4°C) are difficult to compare with freeze-thawed cells (37°C), particularly related to temperature-sensitive protein functionality, some of our findings indicate that S-phase collapse of replication forks may contribute to cold-induced DNA damage. For instance, this mechanism may explain the low levels of exclusive DSB in cells cooled between 8°C and 24°C. In addition, 53BP1 staining in cold-preserved kidney is strongly localized in tubules, the site with the highest proliferation rate 45 .
Our study further documents the absence of robust DNA repair following rewarming, which seems related to the lasting failure of recovery of ATP production. DSB can be maintained for a long time and repaired by ATP-mediated pathways 46 . However, in the continuous presence of ROS and lack of ATP 25 , cells cannot complete DNA repair 47 . Collectively, the substantial prevalence of cooling-induced DNA damage and subsequent lack of repair may explain previous observations documenting the arrest of the cell cycle following cooling and rewarming48,49.
In addition to cells, our data also substantiate cold-inflicted DNA damage in the clinical setting of transplantation, ie, during a standard static deep cooling of kidney in UW preservation solution. Similar to cells, DNA strand breaks in kidney, as evidenced by PFGE, increase with longer duration of storage at 4°C and coincide with increased levels of ROS. The timing of DNA damage is in agreement with the recommended 18 h limit of cold ischemia time for kidney preservation 50 , but also with observations that preservation exceeding 6 h is associated with increased acute rejection, delayed graft function, and decreased graft survival in human kidneys51–53. While prolonged cold storage of kidney transplants adversely affects clinical outcome54,55, the question is to what extent DNA damage contributes. First, extensive and persistent DNA strand breaks may initiate cell cycle arrest, leading to premature senescence 56 through expression of p21CIP1/WAF1 and p16Ink4a 57,58 and induction of a senescence-associated secretory phenotype (SASP) with prominent expression of major pro-inflammatory cytokines, such as interleukin-1α (IL-1α), IL-1β, IL-6, and IL-8. Through SASP, senescence may set the stage to obstruct tissue repair and promote graft immunogenicity. Alternatively, the DNA damage contracted during cooling may lead to genomic instability. Indeed, cancer is a major cause of morbidity and mortality in renal graft recipients. When compared with the general population, kidney transplant recipients have a six- to eightfold higher incidence of renal cell carcinoma59,60. However, the vast majority of cancer cases occurs in native kidneys of the recipient left in situ, amounting nearly 90%. Possibly, this reflects the long-term exposure of kidney transplant recipients to many additional factors promoting tumorigenesis, including metabolic derangement due to end-stage renal failure prior to transplantation, the use of immunosuppressive drugs, and a higher incidence of infections59,61,62. Collectively, our data suggest that cold-inflicted DNA damage bears primarily on graft function and survival by disturbing cellular function, rather than increasing cancer risk in the graft.
The current study uncovers the deleterious effect of cooling, without the need of rewarming, on DNA integrity. Given that DNA repair is erroneous by nature, cooling-inflicted DNA damage may affect cell survival, proliferation, and genomic stability, thus significantly impacting cellular and organ function. DNA integrity may be preserved by avoiding deep and prolonged cooling or by treatment with selected compounds, with relevance to the laboratory setting, cell storage, and transplantation procedures.
Supplemental Material
sj-docx-1-cll-10.1177_09636897221108705 – Supplemental material for Cooling of Cells and Organs Confers Extensive DNA Strand Breaks Through Oxidative Stress and ATP Depletion
Supplemental material, sj-docx-1-cll-10.1177_09636897221108705 for Cooling of Cells and Organs Confers Extensive DNA Strand Breaks Through Oxidative Stress and ATP Depletion by Marziyeh Tolouee, Koen D. W. Hendriks, Fia Fia Lie, Lucas P. Gartzke, Maaike Goris, Femke Hoogstra-Berends, Steven Bergink and Robert H. Henning in Cell Transplantation
Supplemental Material
sj-docx-2-cll-10.1177_09636897221108705 – Supplemental material for Cooling of Cells and Organs Confers Extensive DNA Strand Breaks Through Oxidative Stress and ATP Depletion
Supplemental material, sj-docx-2-cll-10.1177_09636897221108705 for Cooling of Cells and Organs Confers Extensive DNA Strand Breaks Through Oxidative Stress and ATP Depletion by Marziyeh Tolouee, Koen D. W. Hendriks, Fia Fia Lie, Lucas P. Gartzke, Maaike Goris, Femke Hoogstra-Berends, Steven Bergink and Robert H. Henning in Cell Transplantation
Supplemental Material
sj-docx-3-cll-10.1177_09636897221108705 – Supplemental material for Cooling of Cells and Organs Confers Extensive DNA Strand Breaks Through Oxidative Stress and ATP Depletion
Supplemental material, sj-docx-3-cll-10.1177_09636897221108705 for Cooling of Cells and Organs Confers Extensive DNA Strand Breaks Through Oxidative Stress and ATP Depletion by Marziyeh Tolouee, Koen D. W. Hendriks, Fia Fia Lie, Lucas P. Gartzke, Maaike Goris, Femke Hoogstra-Berends, Steven Bergink and Robert H. Henning in Cell Transplantation
Supplemental Material
sj-docx-4-cll-10.1177_09636897221108705 – Supplemental material for Cooling of Cells and Organs Confers Extensive DNA Strand Breaks Through Oxidative Stress and ATP Depletion
Supplemental material, sj-docx-4-cll-10.1177_09636897221108705 for Cooling of Cells and Organs Confers Extensive DNA Strand Breaks Through Oxidative Stress and ATP Depletion by Marziyeh Tolouee, Koen D. W. Hendriks, Fia Fia Lie, Lucas P. Gartzke, Maaike Goris, Femke Hoogstra-Berends, Steven Bergink and Robert H. Henning in Cell Transplantation
Footnotes
Author Contributions
Conceptualization: S.B. and R.H.H; methodology: M.T., K.D.W.H., F.H-B., M.G.; formal analysis: M.T., K.D.W.H., F.F.L., R.H.H.; investigation: M.T., K.D.W.H., F.H-B., M.G, L.G., F.F.L.; writing—original draft: R.H.H.; writing—review and editing: M.T., K.D.W.H., S.B., L.G, and R.H.H; visualization: M.T., K.D.W.H., R.H.H.; supervision: S.B and R.H.H.; funding acquisition: R.H.H.
Ethical Approval
Ethical Approval is not applicable for this article.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects. Animal tissue was procured from the slaughter house.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Consent to Publish
All of the authors have consented to publish this research.
Data Availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: R.H.H. is member of the Scientific Advisory Board and has received honorarium to employer from Sulfateq B.V., a company that holds patents on hydroxychromanols. Sulfateq provided compounds free of charge. No research funding was provided and Sulfateq was not involved in the design of the study, nor in collection, analysis, and interpretation of data and writing of the article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support was received from the University Medical Center Groningen (MD/PhD grant to K.D.W.H.) and the European Space Agency (Research agreement collaboration 4000123556 to R.H.H.).
Funding bodies were not involved in the design of the study and collection, analysis, and interpretation of data and in writing the article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.