
Abstract
Allogeneic stem cell transplantation is a cure for patients suffering from thalassemia major (TM). Historically, patients were limited by the selection of donors, while the advancement of haploidentical stem cell transplantation (haplo-SCT) has greatly expanded the donor pool. However, the outcomes of haplo-SCT in TM recipients vary between different programs. In this study, we retrospectively studied 73 pediatric TM patients (median age, 7 years; range, 3 to 14 years) who underwent haplo-cord transplantation. Both the estimated overall survival and transfusion-free survival were 95.26% (CI 95.77% to 96.23%). Neither primary nor secondary graft failures were observed. The median follow-up period was 811 days (range, 370 to 1433 days). Median neutrophil and platelet engraftment times were 22 days (range, 8 to 48 days) and 20 days (range, 8 to 99 days), respectively. Acute graft-versus-host disease (aGVHD) was observed in 52% of patients and of these, 25% developed grade III to IV aGVHD. Cord blood engraftment was associated with delayed immune recovery and increased aGVHD severity. Viral DNAemia occurred in a relatively high proportion of patients but only 7% of patients developed CMV disease, while another 7% of patients had post-transplantation lymphoproliferative disorder. Long-term complication outcomes were good. Only one patient developed extensive chronic GVHD. No surviving patients were reliant on blood transfusion by the time this manuscript was submitted. This is one of the largest studies on the outcomes of pediatric TM patients who received stem cell transplantations from alternative donors. The haplo-cord program is safe and practical for TM patients that do not have matched donors.
Introduction
Allogeneic hematopoietic stem cell transplantation (HSCT) provides a potential cure for thalassemia major (TM) patients and it is a more cost-effective treatment than lifelong blood transfusion and chelation therapy 1 –3 . Transplants from matched related or unrelated donors are the primary treatment choice for individuals with TM and other hemoglobinopathies 4 . However, less than a quarter of patients in China find fully human leukocyte antigen (HLA)-matched unrelated donors 5,6 . In the last decade, haploidentical stem cell transplantation (haplo-SCT) has multiplied the options for alternative donors. Recent advances in haplo-SCT programs, particularly the optimization of graft selection strategies, the development of in vivo and ex vivo T cell depletion methodologies, and the application of post-transplantation cyclophosphamide (PTCY), have greatly improved overall survival (OS) and event-free survival (EFS), as well as reducing comorbidities 7 –12 .
However, studies investigating the application of haplo-SCT for TM patients are limited. Early attempts have shown an association between TM recipients, a high graft failure rate, and a low probability of survival 10,13,14 . Nevertheless, recent haplo-SCT protocols have exhibited competitive or even superior outcomes, with OS and EFS values up to 96% of those using matched related and unrelated donors 11,15 –18 . However, the outcomes of TM patients that received haplo-SCT vary between different transplant programs. Therefore, additional evidence to confirm long-term safety and efficacy is required.
The current study developed a co-transplantation program for pediatric TM patients consisting of haplo-SCT and a single-dose transfusion of unrelated cord blood on day 6 after transplantation (haplo-cord program) 19,20 . As the haplo-cord program has shown promising results in other diseases 20 –24 , we hypothesized that this program would have encouraging outcomes and long-term advantages in patients with TM.
Subjects and Methods
Patient Characteristics
We retrospectively reviewed the data for 73 TM patients (27 females and 46 males) aged 3 to 14 years (median age 7 years) who had undergone haplo-cord transplantation at Shenzhen Children’s Hospital, China between September 2016 and June 2019. One patient who received haplo-cord transplantation as a second transplantation was excluded before the analysis. All patients, donors, and their respective patient and donor parents were given written consents for the collection, analysis, and publication of their outcome data. This study was approved by the Institutional Review Board of Shenzhen Children’s Hospital in accordance with the Helsinki Declaration.
TM patients in this study included those diagnosed with β-TM or compound β-TM and α-thalassemia based on a genetic examination. Risk assessments were performed based on age at transplantation, serum ferritin level, and hepatosplenomegaly severity 25 . Patients were classified into low-risk, intermediate-risk, and high-risk groups before transplantation.
Haploidentical Donor Selection
We performed high-resolution HLA typing for HLA-A, B, C, DR, and DQ for all donors and recipients. Selection of non-maternal haploidentical donors was favored unless a maternal donor was the only available option. Granulocyte colony-stimulating factor (GCSF, 10 μg/kg/d) was administered to donors once a day for 4 consecutive days for stem cell mobilization before transplantation. Peripheral blood stem cells (PBSCs) with or without bone marrow (BM) were collected from haploidentical donors. Apheresis started on day 5 after mobilization and, if required, continued on the next day until at least 20 × 107 nucleated cells/L/body weight of the recipient were collected.
Cord Blood
Cord blood was selected based on both HLA typing and total nucleated cell counting as shown in previous published strategies by others 19,26 . Grafts that matched for at least four of six HLA loci (HLA-A, HLA-B, and HLA-DR) and contained a minimum nucleated cell count of 1 × 107/kg prior to freezing were selected.
Preparative Regimen
Pre-transplantation immunosuppressive therapy included oral hydroxyurea (30 mg/kg/d) and oral azathioprine (3 mg/kg/d) once a day for at least 90 days prior to transplantation. The conditioning regimen consisted of 50 mg/kg/d cyclophosphamide (CY) for 2 days (days -8, -7) followed by 40 mg/m2/d fludarabine (FLU) for 5 days (days -6, -5, -4, -3, -2), and 2.8 to 3.6 mg/kg/d busulfan (BU) for 3 days (days -6, -5, -4). BU dose was determined based on the risk assessment status. It was adjusted by testing the serum BU level to maintain a steady-state concentration between 600 and 800 μg/L 17 . Rabbit anti-thymocyte globulin (r-ATG, 1 mg/kg/d) was administered for 3 days (days -3, -2, -1). Thiotepa (TT, 10 mg/kg/d) was administered to 61 patients on day -3 (Supplemental Fig. S1). Notably, only three of the first 15 recipients received TT due to medication availability.
Post-Transplantation Immunosuppression
GVHD prophylaxis included PTCY (50 mg/kg/d, days +3 and +4) and a short course of methotrexate (10 mg/m2/d on day +7, then 7 mg/m2/d on days +9 and +12). Tacrolimus (0.04 mg/kg/d) was administered via continuous intravenous infusion over 24 h from day +5 until engraftment or until the patient was able to take oral prescriptions. Tacrolimus administration was targeted to maintain a level between and 5 to 15 ng/mL until day +180 and was then tapered unless there was evidence of acute GVHD (aGVHD) or chronic GVHD (cGVHD). Mycophenolate mofetil (MMF, 30 mg/kg/d) was administered intravenously every 8 h from day +6 onward and changed to an oral prescription until day +30. MMF was tapered after day +30 until day +60 unless signs of GVHD presented.
Supportive Care
Patients were treated in isolated units with high-efficiency particulate-free air filters under strict regulation. From day 8, all patients received an infusion of heparin (150 unit/kg/d) over 20 h and oral ursodeoxycholic acid (12 mg/kg/d) until +20 and +90 days, respectively, unless complications occurred. All patients received empiric broad-spectrum antibiotics to prevent septicemia until the resolution of neutropenia if there was no evidence of bacterial infection. Oral posaconazole (4-6 mg/kg/d) was administered as an anti-fungal prophylactic from day +3 to day +180 unless a fungal infection was evident. Oral sulfamethoxazole (25 to 50 mg/kg/d) was administered three days per week if the total white blood cell count exceeded 3 × 109/L until at least +90 days to prevent pneumocystis.
Patients were screened weekly for cytomegalovirus (CMV) and Epstein-Barr virus (EBV) viremia using a real-time PCR-based method until day +120, then at every return visit. Pre-emptive antiviral therapy was initiated if patient plasma CMV-DNA levels exceeded 1000 copies/ml or levels exceeded 400 copies/ml in combination with highly suspect symptoms of CMV disease. Ganciclovir was used as first-line treatment. Foscarnet was administered to patients suspected or confirmed to have drug-resistant CMV variant mutations. Patients with rising EBV-DNA copy numbers or lower levels of copy numbers with clinical evidence of a post-transplantation lymphoproliferative disorder (PTLD) were considered for intervention. Discontinuation of immunosuppressants was the first consideration. Rituximab (375 mg/m2) per week was administered for up to 28 days in patients with symptom progression. PTLD diagnosis was confirmed based on biopsies and radiological findings. Cytotoxic T lymphocyte (CTL) therapy against CMV or EBV was used in patients with a poor response to conventional treatment.
Patient Evaluations
All patients received high-volume blood transfusions at least 180 days prior to haplo-SCT to maintain a hemoglobin level over 100 g/L. Chelation therapy was conducted according to standard guidelines and a course of high-intensity chelation therapy consisting of a continuous infusion of deferoxamine over 18 h per day was applied to patients over four consecutive days to maintain a target serum ferritin level below 1000 ng/mL before transplantation. Donor-specific anti-HLA antibodies were assessed in patients who received haplo-SCT after January 2018. Patients with high donor-specific anti-HLA antibody titres were advised to change donors or receive treatment to reduce antibody titres as per the institutional protocol if there were no other available donors. Graft chimerism was assessed using a short-tandem-repeat (STR) polymerase chain reaction assay.
Definition of Engraftment
Myeloid engraftment was defined as the first day the absolute neutrophil count (ANC) exceeded 0.5 × 109/L for three consecutive days. Platelet engraftment was defined as the first day platelet count exceeded 20 × 1012/L for seven consecutive days. Primary graft failure was defined as an ANC below 0.5 × 109/L, hemoglobin below 80 g/L, and platelets below 20 × 1012/L by day +28. Secondary graft failure was defined as an ANC below 0.5 × 109/L after initial engraftment excluding primary disease, infection, or drug toxicity. Primary poor graft function (PGF) was defined as bilinear severe cytopenia with or without transfusion requirements after day +28 (which occurred in a situation of full-donor chimerism). Secondary PGF was defined as (1) severe cytopenia with two or three lines that developed after cell engraftment and lasted for over 30 days; (2) myelodysplasia (which occurred in a situation of full chimerism); and (3) excluding other causes of cytopenia 27,28 .
Statistical Methods
OS and transfusion-free survival (TFS) were calculated using the Kaplan-Meier estimator using GraphPad Prism (version 8) software. Patients that were alive and had not experienced any further events by August 31, 2020 were censored.
Results
Patient Characteristics
Patient characteristics are shown in Table 1. The median age was 7 years (range, 3 to 14 years) and the female: male ratio was 0.6. Haploidentical donors were the fathers, mothers, siblings, and other relatives of the recipient in 54%, 11%, 24%, and 1% of cases, respectively. Twenty-three (32%) of patients matched the ABO blood type of their donors. Recipients were matched to their donors for 5 (63%), 6 (21%), 7 (12%), and 8 (5%) HLA loci. Sixteen patients (22%) with an ABO blood type matching their haploidentical donors received both BM and GCSF-mobilized PBSCs. The remaining patients (78%) received only PBSCs from haploidentical donors.
Patient Characteristics.
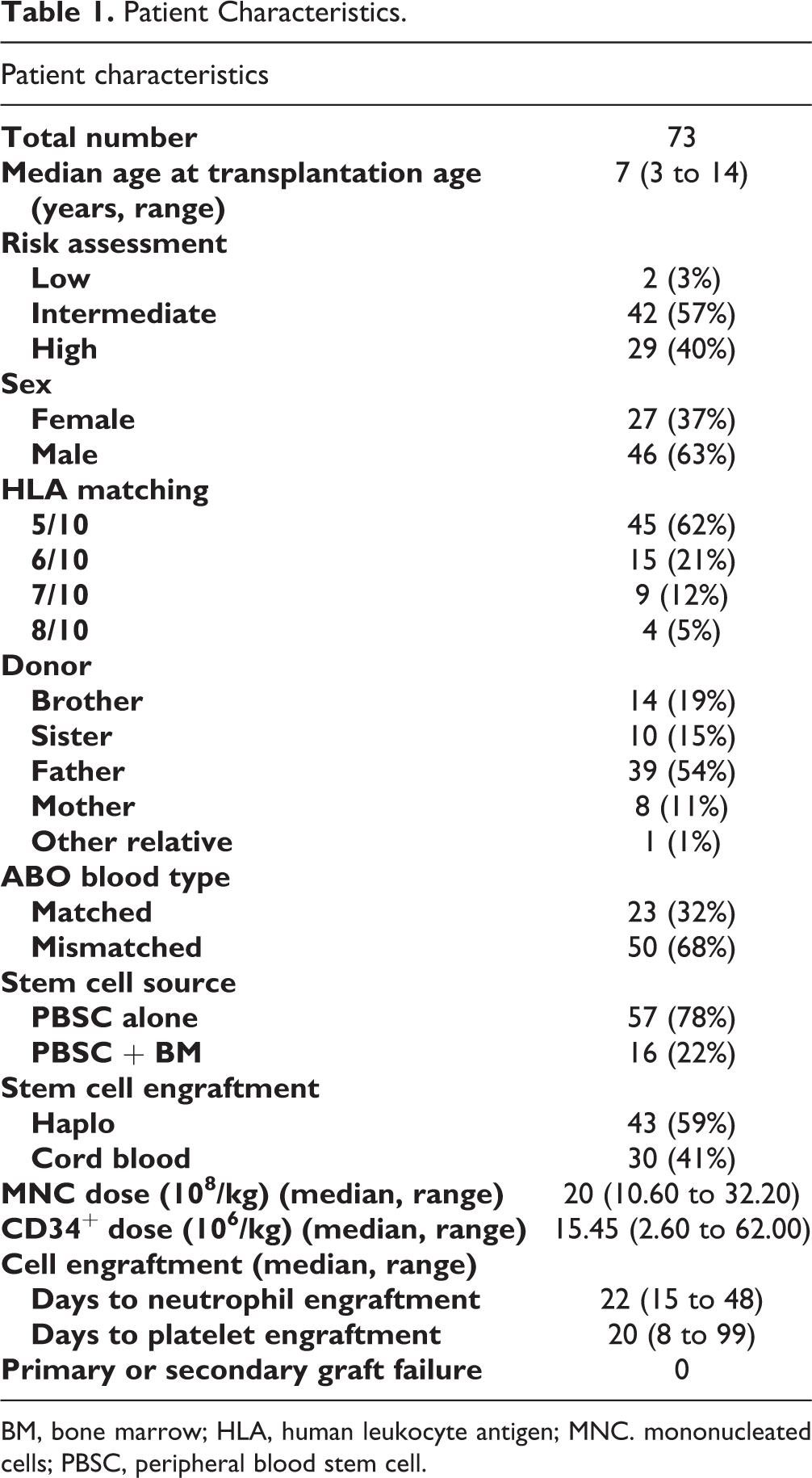
BM, bone marrow; HLA, human leukocyte antigen; MNC. mononucleated cells; PBSC, peripheral blood stem cell.
No primary or secondary graft failure was observed. Forty-three (59%) patients were engrafted with haploidentical grafts alone. The others (41%) were engrafted with cord blood or had mixed chimerism of both types of grafts. Notably, all patients with mixed chimerism of both haploidentical grafts and cord blood following the transplant eventually reached full engraftment of cord blood (STR > 99%) by 12 months after transplantation (Supplemental Fig. S2). The median CD34+ cell number of haploidentical grafts was 18.52 × 106/kg (range, 2.6 to 62 × 106/kg). The median time to myeloid and platelet engraftment was 22 days (range, 8 to 48 days) and 20 days (range, 8 to 99 days), respectively.
GVHD and Other Complications
As shown in Table 2, 20 (27%) patients developed grade I to II aGVHD and 18 (25%) patients developed grade III to IV aGVHD. Eighteen of these patients received steroids against aGVHD in addition to standard GVHD prophylaxis. Twenty-seven (37%) patients developed cGVHD, but only one patient developed extensive cGVHD (grade III to IV).
GVHD and Other Complications in Haplo-Cord Transplant Patients.

GVHD, graft versus host disease; CMV, cytomegalovirus; EBV, Epstein-Barr virus; PTLD, post transplant lymphoproliferative disease; HHV6, type 6 human herpesvirus; BKV, BK virus; PGF, poor graft function; SOS, sinusoidal obstruction syndrome; VOD, veno-occlusive disease.
Cytopenia was observed in 10% of patients. Four (5%) and two (3%) patients developed primary and secondary PGF, respectively. Eighteen patients (25%) developed grade III to IV mucositis. Six patients (8%) developed sinusoidal obstruction syndrome (SOS; previously known as veno-occlusive disease, VOD). Thirty-five (48%) patients had CMV-DNAemia (plasma CMV-DNA > 400 copies/ml) before day +100, but only five (7%) developed CMV disease. Nineteen (26%) patients had EBV-DNA and five (7%) developed PTLD. In addition, four patients with CMV infection and five with EBV infection received CTL therapy because they had a poor response to conventional treatment. Seven (9%) patients developed BK-virus-related hemorrhagic cystitis. Herpes zoster reactivation was observed in seven patients (10%) and one patient developed type 6 human herpesvirus (HHV6) encephalopathy. Four patients (5%) had septicaemia. Uncommon bacterial infections included one case of streptococcal meningitis and another case of systemic tuberculosis. Invasive fungal infections were observed in seven patients (10%). No patients in this report died of a severe infection.
Overall Survival and Transfusion-Free Survival
The estimated OS and TFS were both 95.26% (CI 95.77% to 96.23%) (Fig. 1). Three patients died during the follow-up. One patient had poor graft function and died of hyperacute intracranial hemorrhage on day +418. The other patient developed severe cGVHD and died of respiratory failure on day +370. The third patient died of refractory autoimmune hemolytic anaemia on day +658. The median follow-up period was 811 days (range, 370 to 1433 days).
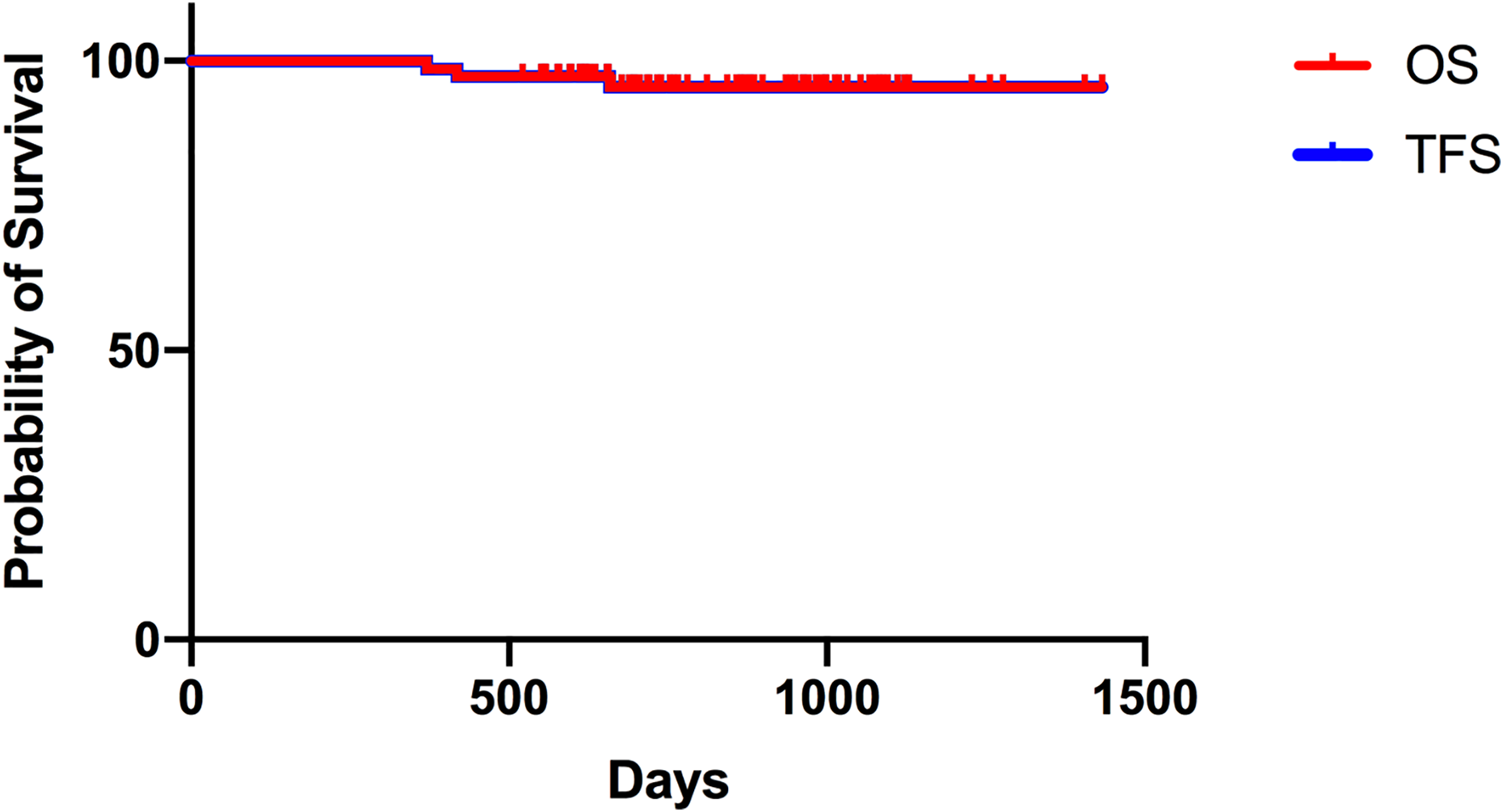
The estimated three-year overall survival (OS) and transfusion-free survival (TFS) for pediatric thalassemia major patients who received haplo-cord transplantations.
Cord Blood Contributed to Delayed Engraftment and Increased aGVHD Severity
Patient outcomes were compared between those engrafted with cord blood and those engrafted with haploidentical grafts (Fig. 2). Patients with cord blood engraftment required more time for neutrophil and platelet engraftment (****p < 0.0001, Mann-Whitney U test). Cord blood engraftment was also associated with a higher risk of developing grade III to IV aGVHD compared to patients engrafted with haploidentical grafts (**** p < 0.0001, Fisher’s exact test) (Fig. 3A). Interestingly, the rates of viral, bacterial, and fungal infections did not differ between patients with haploidentical grafts and cord blood engraftment (Fig. 3B). These two groups of patients did not differ in terms of survival and other complications after transplantation (data not shown).
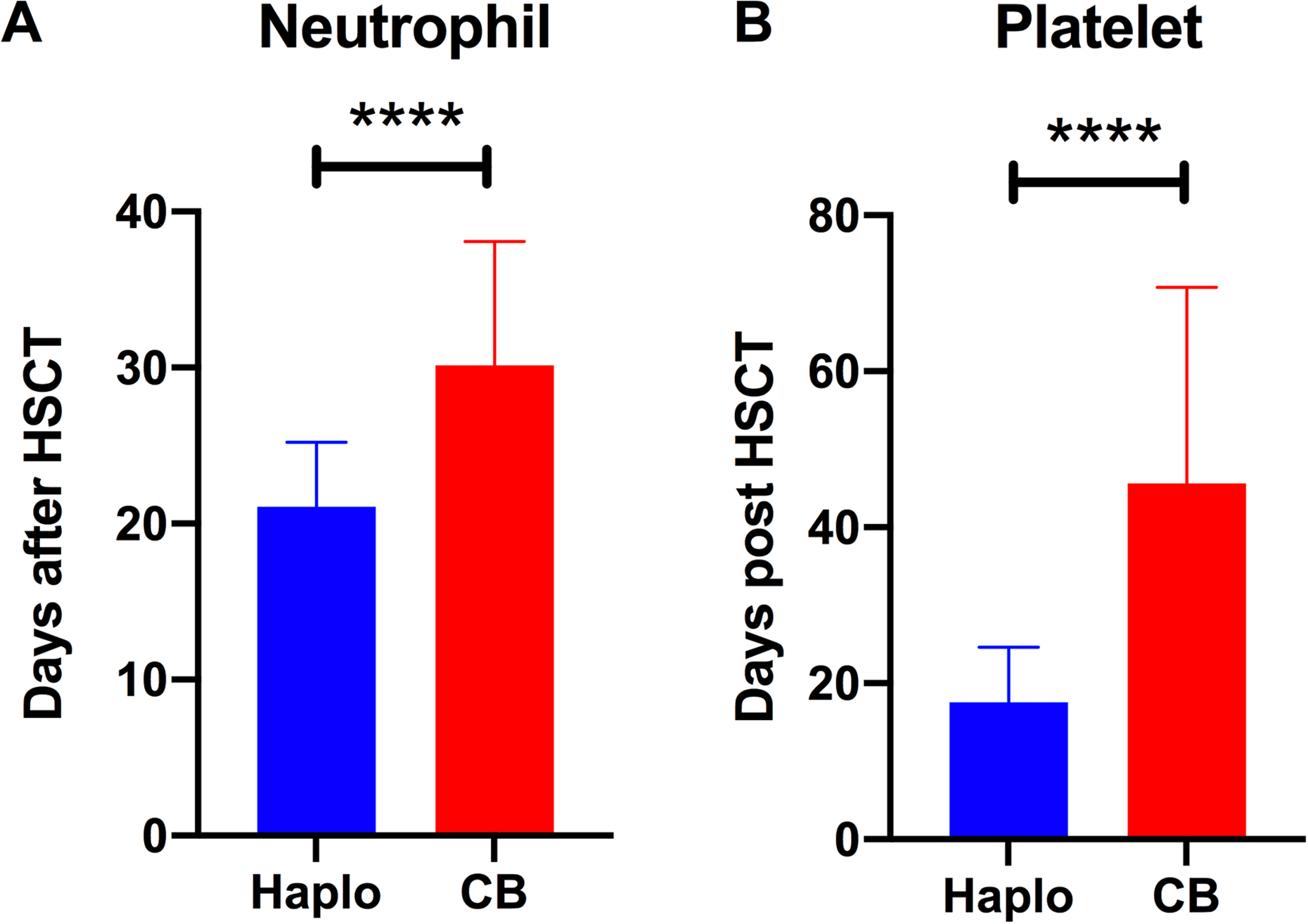
Engraftment of patients that received haploidentical grafts or cord blood. (A) Neutrophil and (B) platelet recovery in patients with haploidentical (Haplo) or cord blood (CB) engraftments (mean ± SD, Haplo vs. CB; (A) 21 ± 4 days vs. 30 ± 8 days and (B) 17 ± 7 days vs. 45 ± 25 days; ****p < 0.001, Mann-Whitney U test).
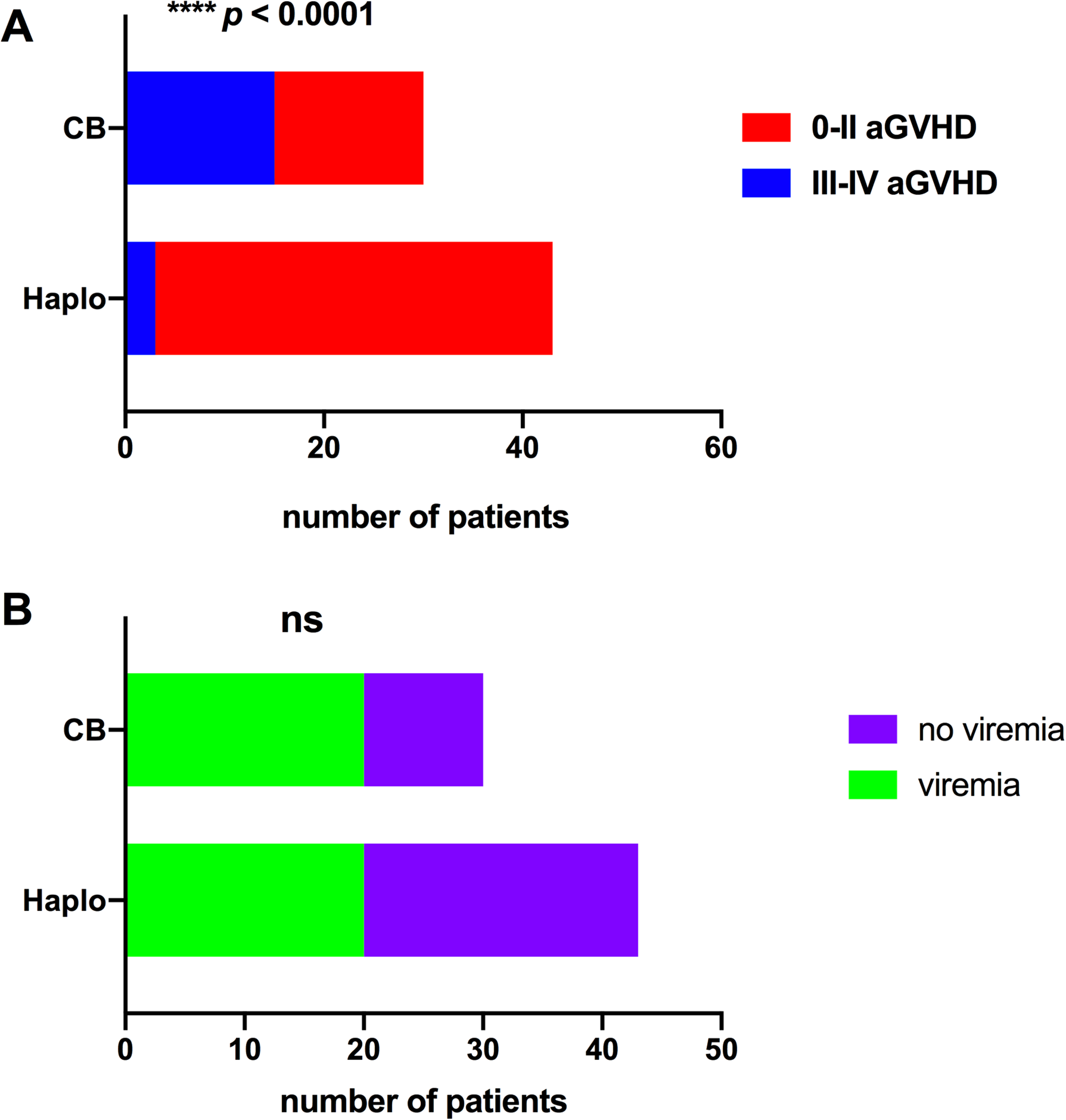
Acute graft-versus-host disease (aGVHD) severity and viral DNAemia rates in patients engrafted with haploidentical grafts (Haplo) or cord blood (CB). (A) The severity of aGVHD in patients with Haplo or CB engraftments (****p < 0.001, Fisher’s exact test). (B) The number of patients with post-hematopoietic stem cell transplantation viral DNAemia in patients that received Haplo or CB engraftments (****p < 0.001, Fisher’s exact test).
Discussion
This is one of the largest studies of pediatric TM patients who received HSCT from alternative donors following a haplo-cord program. Patient outcomes in this study were outstanding, with both OS and TFS rates above 95% and no graft failure. According to the literature, the long-term OS and EFS for TM patients who undergo HSCT from related and unrelated donors ranges from 50% to over 90%, respectively 29,30 . Recent attempts have modified the haploidentical transplant program to reduce the intensity of chemotherapy and to manipulate haploidentical grafts and show encouraging outcomes in thalassemia patients 10,15 –17 . However, these studies are based on small cohorts, have short follow-up periods, or include ex vivo procedures that are not feasible for most transplant centres in China 13,30 . This study describes an alternative approach to cure TM with promising outcomes.
Most TM patients in this study were classified as high-risk patients and considered to have a greater risk of developing drug-toxicity-related complications, particularly severe hepatobiliary complications such as SOS 25,31,32 . We chose to use a myeloablative conditioning regimen in the current protocol because TM patients had very active hematopoiesis in the BM and were more likely to experience graft failure 13,14 . Many transplant centers now use treosulfan as a lower toxicity substitute for BU 33,34 . In addition, other approaches using alternative drugs in place of cyclophosphamide, and using reduced intensity chemotherapy instead of myeloablative chemotherapy have also been introduced in TM recipients of matched related and unrelated donors 35 –37 . This information can help to guide the modification of the current protocol in the future. Nevertheless, despite an intense combination of preparation regimens consisting of alkylators (CY, BU, TT, and FLU) being used in this study, the overall rates of drug-related complications were low and no patients experienced inevitable consequences after treatment.
As a part of GVHD prophylaxis, PTCY was used on days +3 and +4 and therefore could have affected haploidentical graft engraftment. Thus, cord blood was provided as a back-up stem cell source on day +6 to ensure engraftment. The percentage of patients with cord blood engraftment or mixed chimerism of both types of grafts was higher than expected because there was a far greater number of cells in the haploidentical graft than that in the cord blood. Patients with mixed chimerism of both the haploidentical graft and cord blood after engraftment eventually showed full cord blood engraftment (STR > 99%). However, the mechanism behind this is unknown. We suspect that PTCY may have damaged the stem cells in the haploidentical grafts 38 . Another hypothesis is that T cells in the cord blood are more naïve than PBSCs, which contain a more heterogeneous population of T cells 23 . Cord blood also contains a higher proportion of regulatory T cells that contribute to improved tolerance and long-term engraftment 39 –41 .
A relatively higher proportion of patients developed CMV and EBV DNA after receiving a transplant. However, the current study considered viral DNAemia if a patient had plasma CMV-DNA or EBV-DNA titres above 400 copies/ml. This threshold is lower than the standards used in most centres 42 . The lower threshold was used to closely monitor the viral titers, and if required, start pre-emptive antiviral therapy early. In addition, the high rates of viral DNAemia were associated with delayed immune recovery due to a high proportion of cord blood engraftment. However, we did not find a significant difference in the rates of either CMV or EBV DNAemia between patients with haploidentical graft and cord blood engraftment. Overall, the percentage of patients who developed CMV disease or EBV-associated PTLD was acceptable compared to those of most previously published data.
The percentages of patients who developed aGVHD and severe aGVHD were higher than expected in patients with cord blood engraftment. We used a low dose of r-ATG (1 mg/kg/d) as the T cell depletion strategy and applied a classic PTCY strategy to intensify the anti-T cell response to prevent GVHD. We also prioritized the selection of non-maternal relatives owing to the higher incidence of GVHD in recipients that received material from maternal donors 43 . Patients with cord blood engraftment were at a higher risk of developing severe aGVHD (grade III-IV). Patients with cord blood engraftment also required more time for cell recovery. Cord blood engraftment is preferred in leukemia patients with a lower chance of relapse because of the graft-versus-leukemia effect 44 –48 . However, this effect is not beneficial for HSCT recipients with non-malignant diseases such as TM. A potential solution is to use a higher ATG dose to enhance the depletion of early active T cells 49 . Although reduced-intensity chemotherapy may help limit haploidentical graft toxicity, it may not be intense enough for an anti-T cell effect to prevent GVHD. GCSF-mobilized PBSCs are also a risk factor for aGVHD because of the high volume of T lymphocytes in the haploidentical graft 50 . The number of CD34+ cells in the haploidentical graft was also correlated with aGVHD severity (not shown). A possible solution is to use BM instead of PBSC, or increase the proportion of BM in patients that received a combination of BM and PBSC because BM is generally considered a graft source with a lower incidence of both acute and chronic GVHD 51 . In the current study, 22% of patients received a combination of GCSF-primed BM and PBSC. However, no differences were observed in terms of the incidence of transplant-related complications and disease outcomes, probably due to the small sample size (not shown). Thus, the problem of whether to use BM alone or to increase the percentage of BM under the current protocol requires further investigations. In addition, ex vivo graft manipulation with depletion of αβ+ T cells or selection of CD34+ cells have shown to have lower rates of aGVHD and viral reactivation in TM patients that received haplo-SCT 52,53 . However, the incidence of graft failure can reach as high as 45% in these studies. The OS and disease-free survival are relatively low compared to studies without graft manipulation and the current data. Therefore, further investigations are required to balance between transplant-related complications and long-term survival and disease outcomes for TM patients.
Compared to other haplo-cord programs performed in malignant diseases 54 , cGVHD management in the current study was promising. In our study, most patients received only PBSCs from haploidentical donors. Although haplo-PBSC recipients have been correlated with a higher incidence of cGVHD compared to BM recipients 49,55 , we are satisfied with the current result; only a single patient developed extensive cGVHD.
Of the patients who did not survive, two were engrafted with cord blood. However, the precise correlation between cord blood engraftment and mortality was not calculated due to the small sample size in this study. Although we are satisfied with the overall outcomes resulting from the use of the current haplo-cord protocol, optimizations are required to minimize preventable complications. Future studies involving larger cohorts and longer terms are necessary to prove long-term efficacy.
Another limitation of this study is that the use of cord blood may burden patients and their families with additional cost, because of the relatively higher rates of aGVHD and viremia. However, the total cost of treatment for TM patients in our center is low (approximately 30,000 US dollars to 60,000 US dollars for the majority of patients) compared to data reported from developed countries 56 . Nevertheless, further proper clinical trial is required to study the cost effectiveness between centers using different haplo-HSCT protocols for TM patients.
In conclusion, the haplo-cord program is effective and safe for treating pediatric patients with TM. The fact that the haplo-cord program may minimize graft failure is a key strength of the current study. However, concerns regarding cord blood engraftment include the higher rates of severe aGVHD and longer period required for immune reconstitution. Further investigation is required to optimize the haplo-cord protocol to improve survival and limit severe complications.
Supplemental Material
Supplemental Material, sj-png-1-cll-10.1177_0963689721994808 - Co-Transplantation of Haploidentical Stem Cells and a Dose of Unrelated Cord Blood in Pediatric Patients with Thalassemia Major
Supplemental Material, sj-png-1-cll-10.1177_0963689721994808 for Co-Transplantation of Haploidentical Stem Cells and a Dose of Unrelated Cord Blood in Pediatric Patients with Thalassemia Major by Xiaodong Wang, Xiaoling Zhang, Uet Yu, Chunjing Wang, Chunlan Yang, Yue Li, Changgang Li, Feiqiu Wen, Chunfu Li and Sixi Liu in Cell Transplantation
Supplemental Material
Supplemental Material, sj-tiff-1-cll-10.1177_0963689721994808 - Co-Transplantation of Haploidentical Stem Cells and a Dose of Unrelated Cord Blood in Pediatric Patients with Thalassemia Major
Supplemental Material, sj-tiff-1-cll-10.1177_0963689721994808 for Co-Transplantation of Haploidentical Stem Cells and a Dose of Unrelated Cord Blood in Pediatric Patients with Thalassemia Major by Xiaodong Wang, Xiaoling Zhang, Uet Yu, Chunjing Wang, Chunlan Yang, Yue Li, Changgang Li, Feiqiu Wen, Chunfu Li and Sixi Liu in Cell Transplantation
Footnotes
Acknowledgments
We thank Prof. Kuang-Yueh Chiang at the Hospital for Sick Children in Toronto, Canada for constantly consulting our patients.
Ethical Approval
This study was approved by the human ethics committee at Shenzhen Children’s Hospital.
Statement of Human and Animal Rights
This study was conducted in accordance with the Helsinki Declaration for human studies.
Statement of Informed Consent
Written informed consents were obtained from the patients’ parents for the collection and publication of clinical data.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Sanming Project of Medicine in Shenzhen (SZSM 201512033), Shenzhen Fund for Guangdong Provincial High-level Clinical Key Specialties (SZGSP012), and Shenzhen Key Medical Discipline Construction Fund (SZXK034).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.