
Abstract
Cell transplantation is expected to be another strategy to treat lysosomal diseases, having several advantages compared to enzyme replacement therapy, such as continuous enzyme secretion and one-time treatment to cure diseases. However, cell transplantation for lysosomal diseases holds issues to be resolved for the clinical field. In this study, we developed a new ex vivo gene therapy platform using a transplant pack, which consists of a porous membrane made of ethylene-vinyl alcohol in the pack-type and spheroids with scaffolds. These membranes have countless pores of less than 0.1 µm2 capable of secreting proteins, including alpha-galactosidase enzyme, and segregating the contents from the host immune system. When the packs were subcutaneously transplanted into the backs of green fluorescent protein (GFP) mice, no GFP-positive cells migrated to the transplanted pack in either autogenic or allogenic mice. The transplanted cells in the pack survived for 28 days after transplantation. When cells overexpressing alpha-galactosidase were used as donor cells for the packs and implanted into Fabry disease model mice, the accumulation of the alpha-galactosidase enzyme was also observed in the livers. In this study, we reported a new ex vivo therapeutic strategy combining macroencapsulation and cellular spheroids with scaffolds. This pack, macroencapsulated spheroids with scaffolds, can also be applied to other types of lysosomal diseases by modifying genes of interest.
Keywords
Introduction
Lysosomes are acidic organelles that dismantle macromolecules using an array of acid hydrolases. Lipids and glycoproteins engulfed by lysosomes are digested by the enzymes, and their remnant is recycled. The defect in a lysosomal enzyme, which causes lysosomal storage diseases (LSDs), results in the accumulation of substrates in the lysosome, consequently leading to cellular dysfunction in tissues and organs where the enzyme significantly processes the substrates. LSDs comprise more than 70 rare monogenic metabolic disorders and collectively affect 1 in 5,000 live births 1 . Although causative genes are identified in major LSDs, how the disease progresses and clinically manifests still cannot be predicted. Lysosomes also play a role as hubs, including nutrient sensing 2 , cell death 3 , and calcium signaling 4 , in contact with other organelles, such as mitochondria and endosomal reticulum 5 , which might result in heterogeneous clinical features.
A long time after the discovery that lysosomal enzymes mediate a process called cross-correction 6 , the development of biologics as enzyme replacement therapy (ERT) for LSDs rapidly emerged with the aid of the enactment of legislation called the Orphan Drug Act in 1983 in the United States, which was followed by many countries 7 . Still, limited biologics to treat specific LSDs are currently available, therefore, there are several essential issues to be solved. The central nervous system (CNS) is very fragile to lysosomal dysfunction, and approximately 70% of LSDs exhibit progressive neurodegenerative pathology in addition to damaged peripheral organs and tissues 8 . The presence of the blood-brain barrier (BBB) hinders current commercial recombinant enzymes from reaching the most critical site for clinical manifestation 9 , the central nervous system, leaving disorders with neurodegenerative pathology untreatable except for a few enzymes that can be intracerebroventricularly administered 10,11 . Another issue is the generation of neutralizing antibodies to therapeutic biologics, especially when infused into patients with a null mutation in question 12 . Solutions for these concerns are to modulate biologics with the addition of peptides to pass over the BBB 13 and to provide the enzyme activity in question to native enzymes through the replacement of amino acids in the binding pocket for the substrate 14 . A platform that is applicable to all enzymes in lysosomes with an adequate profitable cost and that can easily transform biologics should be developed.
Even in the era of ERT for LSDs, gene therapy has been intensively investigated in both in vivo and ex vivo schemes 15 because of the advantage that one-time treatment could achieve remission, in contrast with ERT, which requires regular injections, usually every other week. Hematopoietic stem cell transplantation (HSC Tx), which was considered to correct enzymatic defects through excreted hydrolases from engrafted hematological cells 16 , partially succeeded in only a limited subpopulation of LSDs, with significant morbidity and mortality 17 , which led to ex vivo gene therapy using autologous HSCs 18 . Ex vivo gene therapy, which fulfills requirements for the platform to enable modifications of enzymes to be transferred, has already been clinically examined for Fabry disease by using autologous hematopoietic stem cells 19 . A concern is a dose of alpha-galactosidase (aGLA) with a mannose-6-phosphate (M6P) moiety to be supplied by exogenous genetically modified hematological cells. In a previous study, we calculated a naïve cell number of 1.7 × 10 12 , which releases aGLA equivalent to clinically used biologics 20 . When patients with Fabry disease received kidney transplantation, cross-correction in other organs, such as the heart, that suffered from aGLA dysfunction was not recognized 21 . Although M6P receptors in the Golgi apparatus of aGLA-overexpressing cells might be saturated enough not to keep aGLA, leading to the release of aGLA with M6P to the circulation, overproduction might affect cellular functions with endosomal reticulum stress in the long term 22 . In the ex vivo gene therapy scheme, we established a system to release aGLA with M6P up to a triple digit increase, to be able to adjust the dose of aGLA to be released and to be exchangeable for quality control.
Islet transplantation, which started with allogenic sources under immunosuppression at the University of Alberta 23 , has paved the way for the field of cell encapsulation technology for transplantation to treat diabetes mellitus 24 . Encapsulation is designed to satisfy the long-term survival of included cells, good glucose sensing and insulin releasing capability, and immunological protection. Methods to encapsulate islets are classified into three categories: nanoencapsulation, microencapsulation, and macroencapsulation, based on the distance of cells from the environment 25 . Layer-by-layer deposition of biomaterials achieved nanoencapsulation of a single cell 26 . An ultrathin polymer membrane that is thicker and more stable than a single cell coating provides microencapsulation for cell clusters 27 . In contrast to these nano or microencapsulation strategies, macroencapsulation devices can be retrieved and monitored upon implantation into subcutaneous tissues. Some devices have already been commercialized, such as Encaptra in ViaCyte, which has proceeded to several clinical trials 28 . The device, which is simply composed of semipermeable membrane vessels and human embryonic stem cell-derived pancreatic progenitors, was subcutaneously implanted as a dose-escalating phase 1/2 study for type I diabetes. Encapsulated cells remained viable for as long as 24 months. We showed that spheroids with scaffolds could be more promising donor materials for transplantation to treat Fabry disease than dispersed cells 20 . To segregate engineered spheroids, macroencapsulation was chosen considering its retrievability when developing neutralizing antibodies following transplantation.
Current treatment strategies for lysosomal diseases are still an impediment, although they have rescued many patients with these disorders. We pursued a novel system to supply a lysosomal enzyme under an ex vivo gene therapy scheme along with tissue engineering technology. We designed it for Fabry disease, which is versatile for all kinds of LSDs, and applied it to LSDs with disturbed CNS pathology.
Materials and Methods
Ethical Approval
Ethical approval to report this case was obtained from the Committee at the Kyoto Prefectural University of Medicine, Japan (M2019-305).
Statement of Human and Animal Rights
All procedures in this study were conducted in accordance with the Animal Care and Use Committee at the Kyoto Prefectural University of Medicine, Japan (M2019-305) approved protocols. This study was accepted by the recombinant DNA experiment committee (2019-63) in Kyoto Prefectural University of Medicine, Japan.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Preparation of immunoisolation membrane pack
Porous immunoisolation membranes made of ethylene vinyl alcohol (EVOH) were provided by Kuraray Co., Ltd. (Tokyo, Japan). The membrane was cut into 2 cm × 1 cm sheets, and two of these sheets with one port were sealed by heating (100°C for 0.5 s) to fabricate an immunoisolation membrane pack.
Scanning Electron Microscopy (SEM) Analysis and Pore Size Measurement
An immunoisolation membrane was washed with acetone and dried overnight at room temperature. A dried sample was immersed in liquid N2 and folded to prepare a cross-section of the membrane coated with a thin layer of Pt-Pd using an ion sputter coater (MC1000, Hitachi High-Tech, Tokyo, Japan). After preparation, the sample was observed by SEM (S-3000 N, Hitachi High-Tech, Tokyo, Japan) at an acceleration voltage of 10.0 kV. Pore sizes in SEM data were measured using the “Set Scale…” and “Analyze Particles…” functions of ImageJ (version 2.1.0/1.53c).
Cell Culture
For mouse embryonic fibroblast (MEF) isolation, the uteri were removed from C57BL/6 mice that were 13.5 days pregnant and washed using phosphate-buffered saline (PBS) (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). The head and visceral tissues were removed from the isolated embryos. Using 2 pregnant mice ranging between 18 and 20 days postcoitum, 1 to 5 × 107 cells could be acquired as primary fibroblasts to be used in further experiments. The remaining tissues were washed using fresh PBS, minced with scissors, and transferred into fresh Dulbecco’s modified Eagle medium (DMEM; FUJIFILM Wako Pure Chemical Corporation) supplemented with 10% FBS and 1% penicillin and streptomycin (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Three days after incubation, the outgrowth cells were trypsinized, collected by centrifugation (200 × g for 5 min), and resuspended in fresh medium. Following the first passage, 1 × 10 6 cells were cultured in 150-mm dishes at 37°C in 5% CO2. In this study, we used MEFs within 5 to 10 passages. The MEFs were cultured in DMEM containing 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin at 37°C in 5% CO2.
For the formation of spheroids with scaffolds (SSs), two-milliliter cell suspensions of MEFs at a density of 1 × 106/ml with 1 mg/ml µ-pieces were poured into an Elplasia multiple-pore hanging drop microplate, which is a 6-well plate with an insert holding 648 pores (MPc500, Kuraray Co., Ltd., Tokyo, Japan). SSs in the microplates were cultured for 2 days and then transferred into the immunoisolation membrane pack. The packs were sealed using a sealer (Z-1 Clip Sealer, Techno Impulse Corporation).
Macroencapsulated Spheroid with Scaffold (MESS) Transplantation
Under general anesthesia using isoflurane (1.5%) (v/v air; Mylan Inc., Tokyo, Japan), C57BL/6 mice were subjected to laparotomy, and the kidneys were exposed. Incisions were made horizontally in the renal capsules at the middle of the ventral aspect. An immunoisolation membrane pack encapsulating SSs (SSs-immunoisolation membrane pack) was transferred under the skin of the back of a Fabry mouse 20 using sterilized tweezers. The capsular incisions were closed by a few stitches with 7-0 proline, and the abdominal wounds were closed with two layers. In this paper, “MESS transplantation” is defined as the transplantation of SS-immunoisolation membrane packs.
Tissue Preparation and Alpha-Galactosidase Activity Measurement
Mice were anesthetized and sacrificed 28 days after MESS transplantation, and all tissues (liver, kidneys, and heart) were removed. The organs were assessed for alpha-galactosidase activity and lyso-Gb3 levels and were subjected to histological analysis. Alpha-galactosidase activity in mouse organs and cells was measured by assaying with an artificial substrate, as described previously 14 . Ten microliters of a tissue homogenate was mixed with the substrate solution comprising 5 mmol/L 4-methylumbelliferyl alpha-D-galactopyranoside as a substrate and 117 mmol/L N-acetyl-D-galactosamine as an inhibitor of alpha-galactosidase B (alpha-N-acetylgalactosaminidase) in 0.1 mol/L citrate-phosphate buffer, pH 4.6, in a 1.5 mL microtube, followed by incubation at 37°C for 30 min. Then, the reaction was stopped by adding 950 µL of 0.2 mol/L glycine buffer, pH 10.7. Then, the released 4-methylumberiferone was measured using a spectrofluorometer (F2700; Hitachi Ltd., Tokyo, Japan) or a microplate fluorescence reader (Tecan Group Ltd., Männedorf Switzerland) at excitation and emission wavelengths of 365 nm and 450 nm, respectively. Lyso-Gb3 in the organs of mice and tissues in mice was measured as described previously 29 . Briefly, Lyso-Gb3 in the organs of mice was separated using high-performance LC. An InertSustain C18 column (30 × 3 mm I.D., 5 μm; GL Science, Tokyo, Japan) was kept at 45°C during the procedure. The flow rate was set at 0.3 ml/min, and the injection volume was 2 μl. Then, lyso-Gb3 in the samples was detected by MS/MS. The multiple reaction monitoring (MRM) conditions were optimized with an automatic MRM optimization function. The calculations for the measurement of Gb3 isoforms and lyso-Gb3 and its analogs were performed using LabSolutions (Shimadzu Corporation, Kyoto, Japan). The amounts of lyso-Gb3 and its analogs were calculated from the ratio of the area of the MS peak for lyso-Gb3 or each lyso-Gb3 analog to that for lyso-Gb3 IS.
Histology and Immunohistochemistry
Tissue samples were fixed with 4% paraformaldehyde (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) and embedded in paraffin. Hematoxylin and eosin (HE) staining was performed according to standard procedures. Apoptosis detection was performed using an ApopTag apoptosis detection kit (S7100, Merck Millipore, Billerica, MA, USA) for TUNEL labeling according to the manufacturer’s instructions.
Alpha-galactosidase and green fluorescent protein (GFP) accumulation were analyzed by immunohistochemistry. Briefly, the sections were incubated with 3% BSA and reacted with anti-aGLA rabbit polyclonal antibodies or GFP polyclonal antibody, Alexa Fluor 488 (Thermo Fisher Scientific, A21311), for 30 min at room temperature with light shielding. Furthermore, aGLA antibody was stained using a rabbit-specific HRP/DAB Detection IHC Detection Kit - Micropolymer (ab236469, Abcam) according to the manufacturer’s instructions. Nuclear staining of fluorescence was performed by 4,6-diamidino-2-phenylindole, dihydrochloride (DAPI, Dojindo Molecular Technologies, Inc.). A technical negative control was stained without primary antibody.
Overexpression of the aGla gene
For the overexpression of aGla in MEFs, the sequence of aGla (NM_013463.2) was inserted into the retroviral vector pMXs containing a puromycin resistance gene (#RTV−012, Cell Biolabs Inc., San Diego, CA, USA). The pMX retroviral vector carrying the aGla gene was transfected into MEFs, and infected cells were selected in the presence of puromycin.
Quantitative Reverse Transcriptase Polymerase Chain Reaction (qPCR)
For qPCR, 100 ng of total RNA was reverse-transcribed using the PrimeScript RT reagent Kit (Takara Bio Inc., Shiga, Japan) and KAPA SYBR FAST qPCR Kit Master Mix (2×) Universal (KAPA BIOSYSTEMS, Boston, MA, USA) according to the manufacturers’ recommendations. qPCR was performed using the CFX Connect Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., CA, USA). All reactions were performed in triplicate. PCR primers for the aGla and Gapdh genes were designed to amplify each cDNA using the sense primer (5′−TCTGTGAGCTTGCGCTTTGT−3′) and the reverse primer (5′−GCAGTCAAGGTTGCACATGAAA−3′) for aGla and the sense primer (5′−TGCGACTTCAACAGCAACTC−3′) and the reverse primer (5′−CTTGCTCAGTGTCCTTGCTG−3′) for Gapdh. Calculations were automatically performed by fluorescent quantitative detection system software (CFX96 system, Bio-Rad Laboratories, Inc.).
Statistical Analysis
The results are expressed as the means ± standard errors (SEs). The statistical significance of differences among groups was evaluated using t-tests and standard Bonferroni correction (P = 0.01) (Prism 8.4.3 software, GraphPad Prism Software Inc., San Diego, CA), and P < 0.05 was considered to indicate significance.
Results
Generation of the MESS System
Macroencapsulated spheroids with scaffolds (MESSs) are packs that encapsulate cellular spheroids with scaffolds in a membrane-like pouch to isolate them from the host immune system (Fig. 1A). We previously reported how to generate massive spheroids with scaffolds by the hanging drop method using MPc500 plates to maintain cell viability even in the center of the spheroids 20 and used the same method in this study. We chose a membrane made of ethylene-vinyl alcohol with respect to biocompatibility, permeability, and rigidity to create a bag for macroencapsulation, which can contain 650 spheroids at maximum, assuming a mouse experiment. Four types of membranes with different water permeabilities and membrane thicknesses were prepared to evaluate the performance to cross macromolecules over the membrane (Fig. 1B). Proteins of alpha-galactosidase enzyme were encapsulated, and the packs were immersed in culture medium. The enzyme exuded from the packs in the culture medium was evaluated over time (Fig. 1C). The enzyme activity reached a peak 2 h after submerging them and gradually decreased thereafter. There was no difference in the alpha-galactosidase enzyme activity among these packs (Fig. 1C). We decided to use Pack 3, which has the thinnest membrane for oxygen/nutrient exchangeability and the lowest water permeability to ensure immunoisolation. When the surface and cross-section of the Pack3 membrane were observed using SEM, numerous small holes existed on the surface of both sides, and droplet-like spaces in the middle of the membrane interconnected with pleural thin channels to both the outer and inner sides (Fig. 1D). This droplet-like structure contributed to improving the fractionation properties. The pore size of the membrane was measured to be 0.083 ± 0.001 µm2 (n = 3600). The area of more than 80% of the total pores was less than 0.2 µm2 (Fig. 1D).
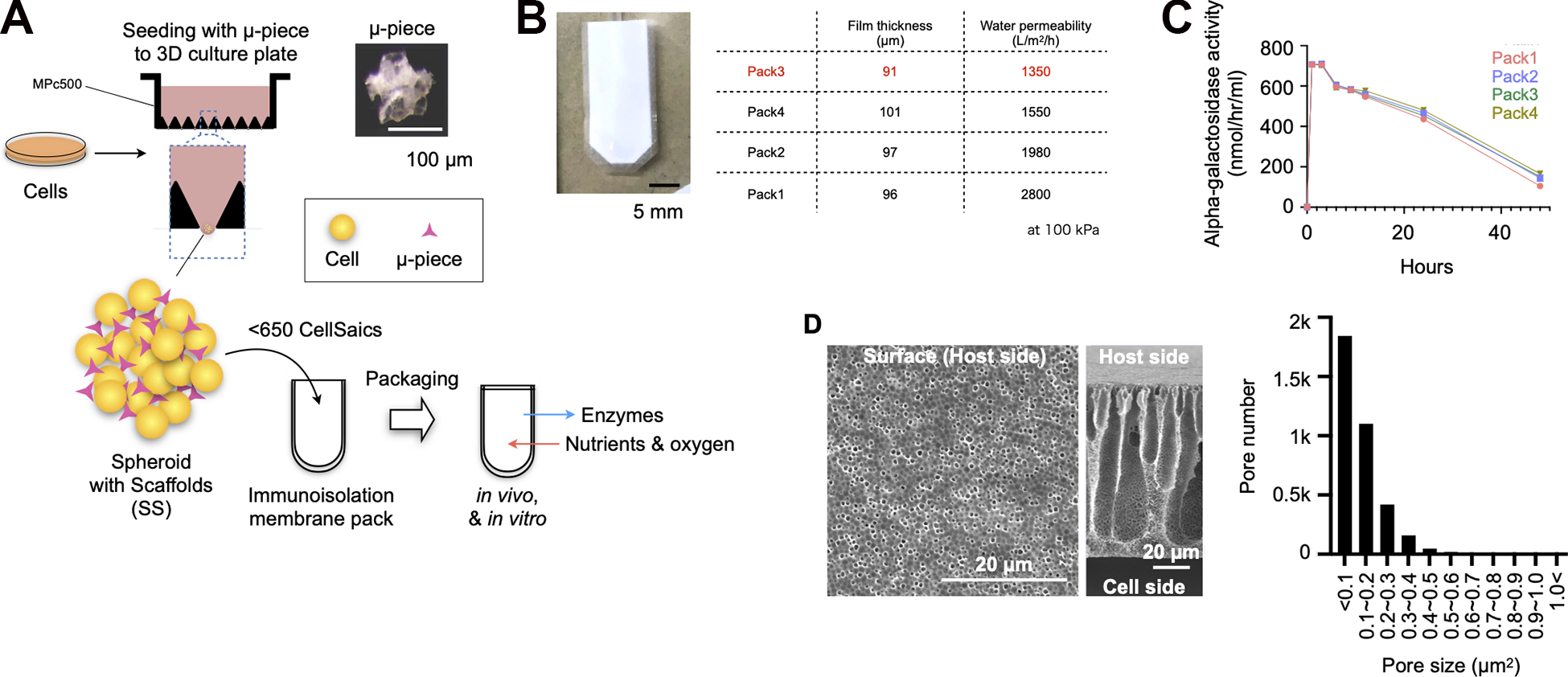
Analysis of the physical properties of the immunoisolated membrane. (A) Illustration of the MESS system. (B) Thickness and water permeability of the immunoisolation membrane. (C) Alpha-galactosidase enzyme activity through the immunoisolation membrane. Alpha-galactosidase enzyme derived from green coffee beans was used. (D) Electron microscopic analysis of the immunoisolation membrane. Graph showing the area of the membrane pores on the host side as a frequency distribution.
Encapsulated Cells in a Pack in Vitro and in Vivo
We chose MEFs as donor cells to encapsulate into the pack because of the ease of culture and gene modifications. To demonstrate the feasibility of encapsulating spheroids with scaffolds (SSs) while maintaining viability and the ability to release lysosomal enzymes, a pack containing approximately 650 spheroids (2 × 106 cells and 2 mg µ-piece) with diameters of 400–500 µm was generated (Fig. 2A, B). SSs and cellular spheroids (CSs), which do not contain scaffolds, were transferred into the packs with culture medium, and they were sealed in a heat fusion machine (Fig. 2B). The packs were incubated in a CO2 incubator while standing against the wall of the culture plate so that SSs or CSs could be easily detected upon histological assessments. After 7 days, either SSs or CSs in the packs were examined by HE and TUNEL staining. The center of the CSs was uniformly a cavity, and many TUNEL-positive cells were present around the cavity (Fig. 2C, D left panels). Alternatively, most cells in SSs were evenly distributed and viable, and TUNEL staining was almost negative (Fig. 2C, D right panels), suggesting that the pack containing SSs can provide a microenvironment to support cell survival and viability for at least 7 days under in vitro conditions.
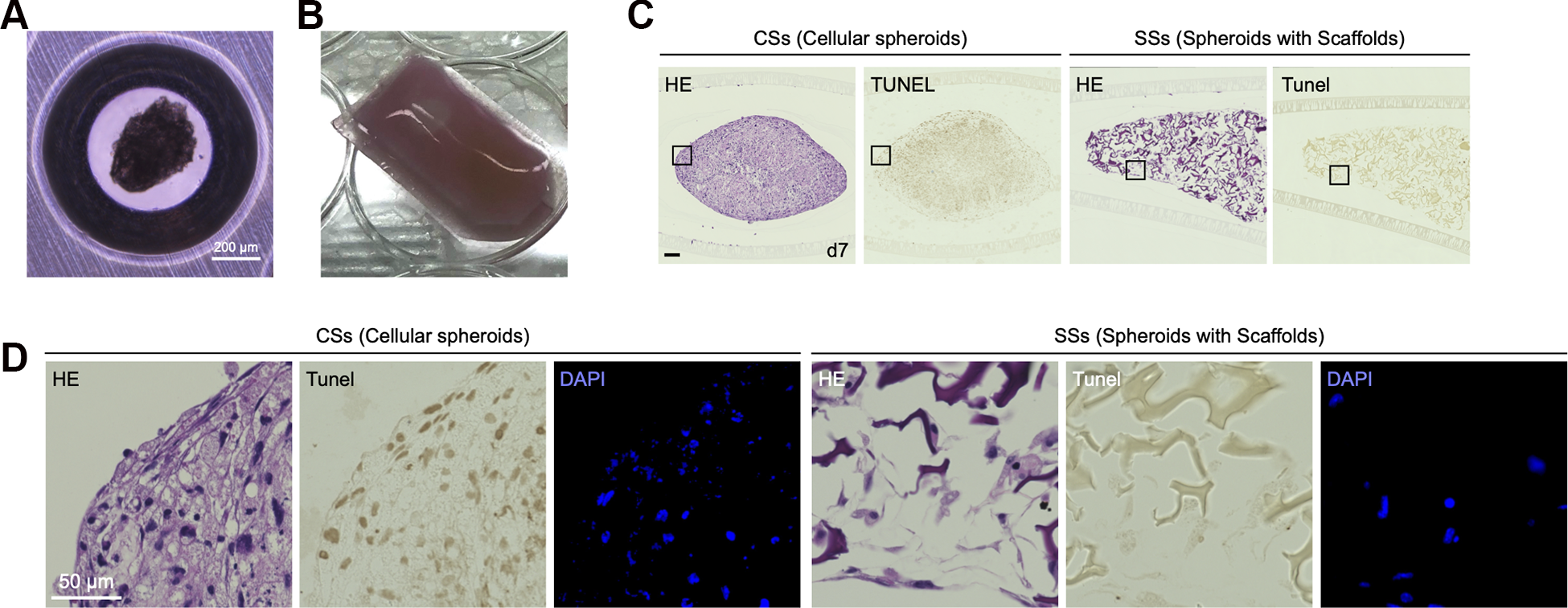
In vitro analysis of cell-encapsulated immunoisolation membranes. (A) SSs formation using MPc500. (B) Immunoisolation membrane encapsulating SSs before transplantation. (C) HE and TUNEL analysis of CSs (without µ-pieces) or SSs (with µ-pieces) encapsulating immunoisolation membranes in vitro for 7 days. Black bar indicates 100 µm. (D) Highly magnified image of the black square in Fig. 2C.
Next, to evaluate whether host-derived cells could invade the packs, they were subcutaneously implanted into the backs of GFP transgenic mice, all cells of which strongly expressed GFP [C57BL/6-Tg (CAG-EGFP)]. The packs containing CSs or SSs were cultured in a CO2 incubator for 24 h before transplantation. The CS-immunoisolation membrane pack formed a thin layer of cell mass on day 7 of transplantation, and there were almost no cells in the pack on day 28 (Fig. 3A). Additionally, we did not identify GFP-positive cells, suggesting that GFP mouse-derived cells cannot migrate through the immunoisolation membrane (Fig. 3B). When the same experiment was performed with SSs, large cell masses were formed both on day 7 and day 28 of transplantation, indicating that the cells were still alive in the immunoisolation membrane pack (Fig. 3C, Fig. S1). No inflammatory cells were observed at the site of transplantation under any conditions (Fig. S1).
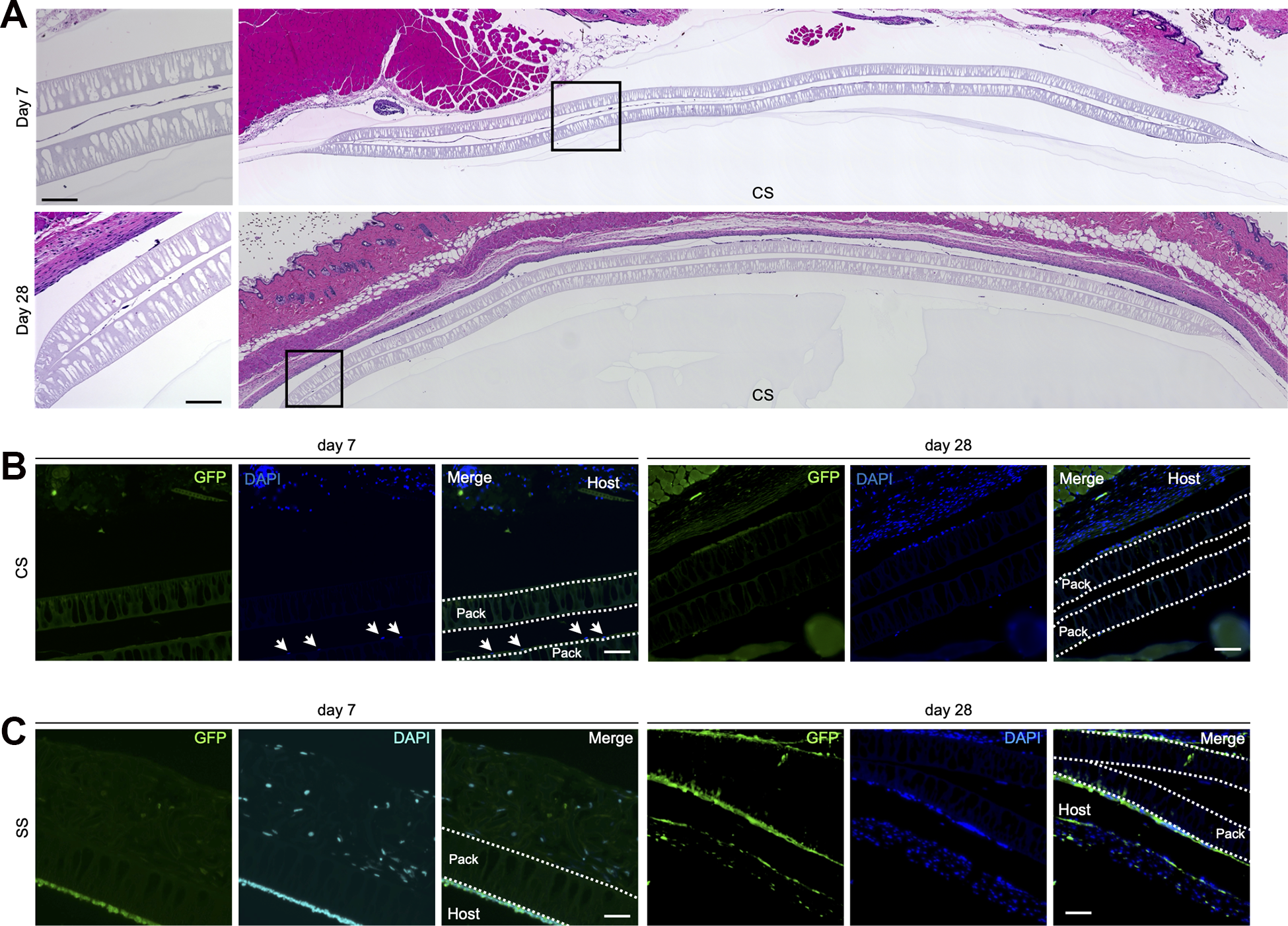
Cell invasion analysis in vivo. (A) HE staining analysis of CS-immunoisolation membrane pack transplanted in GFP mice (C57BL/6 background) for 7 days and 28 days. The transplanted cells were MEFs derived from wild-type C57BL/6 mice. Black bar indicates 100 µm. (B) Fluorescence staining analysis of CS-immunoisolation membrane pack transplanted in GFP mice (C57BL/6 background) for 7 days and 28 days. The transplanted cells were MEFs derived from wild-type C57BL/6 mice. White bar indicates 50 µm. White arrows indicate the nucleus. (C) Fluorescence staining analysis of SS-immunoisolation membrane pack transplanted in GFP mice (C57BL/6 background) for 7 days and 28 days. The transplanted cells were MEFs derived from wild-type C57BL/6 mice. White bar indicates 50 µm.
MESS Transplantation in Autogenic and Allogenic Conditions
To confirm the survival and functionality of the cells enclosed in the pack upon in vivo transplantation, we transplanted the packs in syngeneic and allogenic combinations. First, SSs were generated with C57BL/6-derived MEFs and scaffolds and then encapsulated in the packs. The packs were subcutaneously transplanted into the backs of mice to avoid torsion. The transplanted packs in syngeneic combination were collected on days 7 and 28, and their cross sections were observed by HE and TUNEL staining. The packs were surrounded by a thin layer containing few fibroblasts on day 7, which was stable without thickening or increasing the number of fibroblasts on day 28, indicating a limited fibrotic reaction. On day 28, the scaffolds were retained to support cells, and a large number of cells were also found in the gaps between the scaffolds (Fig. 4A). No inflammatory cells were observed both around and in the packs on either day 7 or day 28. TUNEL staining of the packs was almost negative, in line with in vitro experiments for 7 days, indicating that encapsulated cells were maintained in the pack for 4 weeks in vivo. (Fig. 4B). Next, the transplant experiments were performed in the allogeneic combination, where the recipients were BALB/c and the donors were C57BL/6. As syngeneic grafting was exhibited, transplanted packs in allogeneic combination provided a microenvironment to keep encapsulated cells viable for 4 weeks (Figs. 4C, D). There were no inflammatory cells both around and in the packs on either day 7 or day 28, suggesting that the packs can offer immunoisolation to segregate encapsulated cells from the host immune system and a restricted fibrotic response to the synthetic membranes.
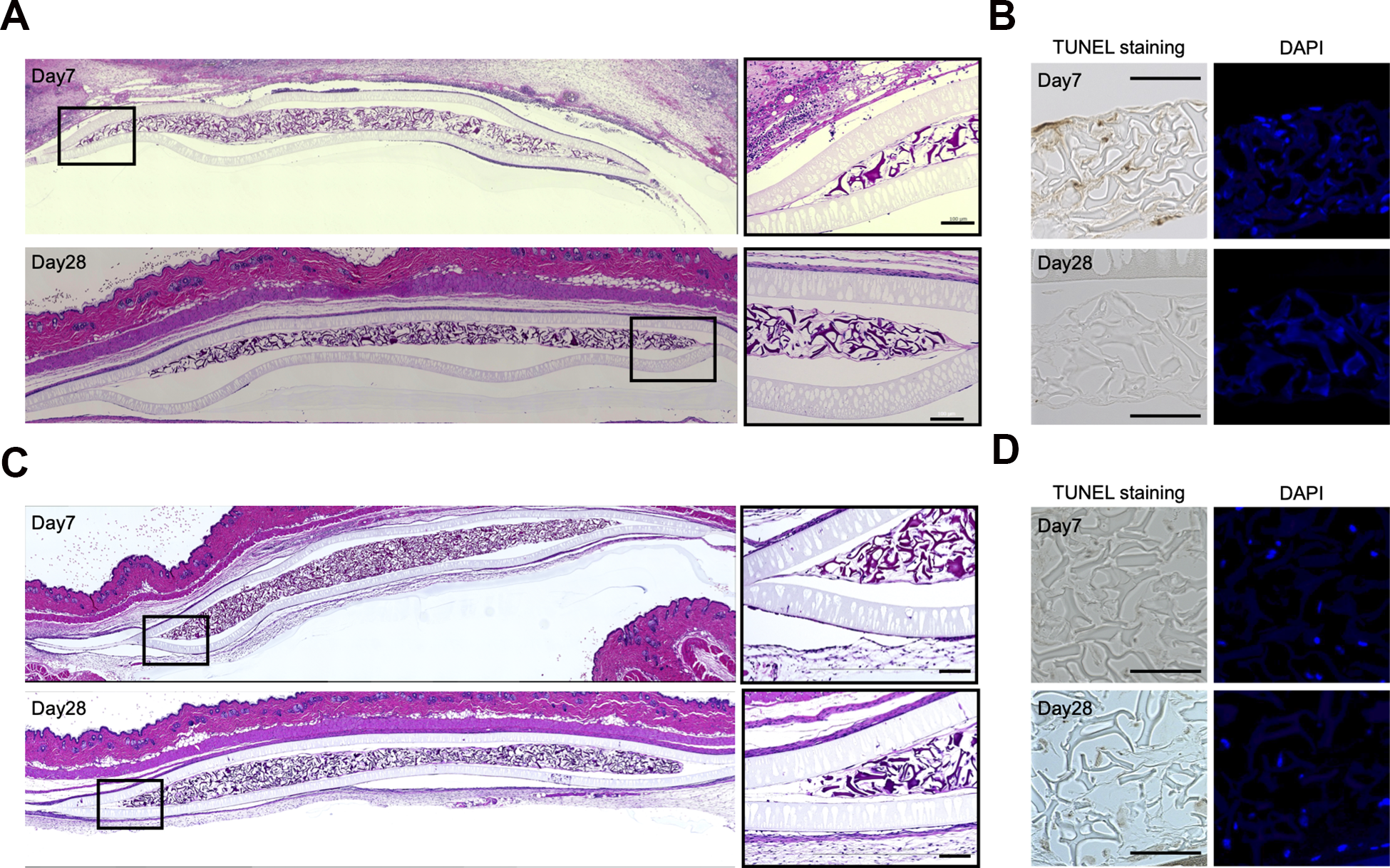
In vivo analysis of the MESS system. (A) HE staining of C57BL/6-derived SS-immunoisolation membrane pack transplanted in C57BL/6 mice. Black bar indicates 100 µm. (B) DAPI and TUNEL staining of C57BL/6-derived SS-immunoisolation membrane pack transplanted in C57BL/6 mice. Black bar indicates 100 µm. (C) HE staining of C57BL/6-derived SS-immunoisolation membrane pack transplanted in BALB/c mice. Black bar indicates 100 µm. (D) DAPI and TUNEL staining of C57BL/6-derived SS-immunoisolation membrane pack transplanted in BALB/c mice. Black bar indicates 100 µm.
The Generation of Genetically Modified Cells to Overexpress Alpha-Galactosidase
A necessary function of donor cells suitable to treat Fabry disease in ex vivo gene therapy schemes is the supra-normal secretion of the alpha-galactosidase enzyme. We constructed pMX-puromycin retroviral vector-based aGla (NM_013463.2) to overexpress alpha-galactosidase (Fig. 5A). MEFs immortalized by SV40 (SV40 1: pBSSVD2005, Addgene plasmid # 21826; http://n2t.net/addgene:21826;RRID:Addgene_21826) were used as donor cells for gene transfer. The MEFs were infected with the recombinant retroviruses and purified by puromycin selection to establish a cell line that stably expresses the aGla gene. The puromycin-selected cells (2 × 105 cells in 6-well plates) showed approximately 180-fold higher aGla gene expression than the original cells (Fig. 5B), and the number of extracellular secreted enzymes in the culture medium was approximately 4-fold higher in monolayer culture (Fig. 5C). Using the established aGla-overexpressing cells, we generated SSs based on the same protocol in the preceding experiments. We analyzed the alpha-galactosidase enzyme activity of the culture medium in the packs to examine the influence of the membrane on the release of the enzymes in vitro. Following incubation of the SS-immunoisolation membrane pack for 2 days in a CO2 incubator, the culture medium was measured (Fig. 5D). The packs released smaller amounts of enzymes than SSs alone (Fig. 5D). Although the membrane partially interfered with the pass-over of macromolecules, SSs constituting aGla-overexpressing cells in the pack showed an approximately 3-fold increase in enzyme activity in the media compared with that released from naïve cells.
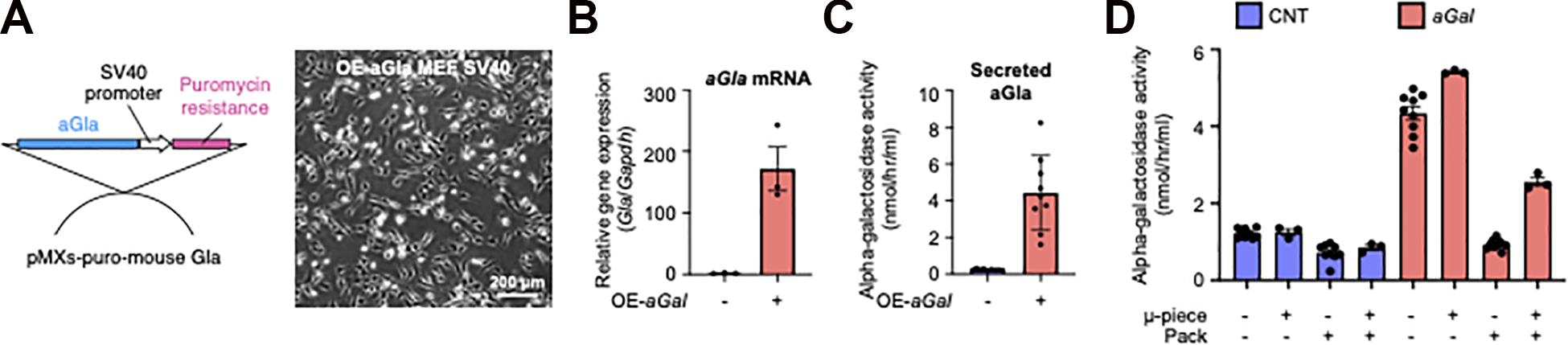
Design of genetically modified cells suitable for encapsulation in the immunoisolation membrane. (A) Design of the overexpression system of the aGla gene using retroviruses. Immortalized MEFs (C57BL/6) with SV40 were used as cells for gene transfer. White bar indicates 200 µm. (B) Analysis of aGla gene expression in aGLA-overexpressing MEFs. (C) Analysis of extracellularly secreted alpha-galactosidase enzyme activity in aGLA-overexpressing MEFs. (D) The value of alpha-galactosidase activity secreted from the SS-immunoisolation membrane pack. aGal: aGal gene overexpression, CNT: cells without any treatment. Pack: Immunoisolation membrane pack.
MESS Transplantation Using Genetically Modified Cells
The packs were transplanted into Fabry disease model mice, and 28 days later, the packs were inspected by HE and TUNEL staining. Similar to unmodified MEFs, a large number of cells were present inside the packs for 4 weeks. Additionally, few TUNEL-positive cells were found (Fig. 6A). Immunohistological analyses using alpha-galactosidase antibody demonstrated that the cells in the device were strongly positive and that the cells around the membrane were also positive. Because the immunofluorescence staining in Fabry disease model mice was completely negative (Fig. S2), the positive staining of cells surrounding the pack indicates uptake of released alpha-galactosidase from the pack graft by host cells (Fig. 6B). From these results, it is expected that encapsulated cells are less likely to undergo apoptosis in the packs and are capable of stable proliferation. Additionally, because no accumulation of cells was observed around the transplanted area, it is supposed that no cells had leaked out the packs. When the alpha-galactosidase enzyme activity levels in the plasma fraction of the blood of the mice transplanted with the packs were evaluated on days 7 and 28, there was a significant increase on day 7 (Fig. 6C). Alpha-galactosidase in the blood is easily taken up by the liver 20 . Therefore, 28 days after MESS transplantation, the liver of the Fabry disease model mouse was stained with alpha-galactosidase antibody, and it was positive in the MESS transplantation group (Fig. 6D). We then measured the alpha-galactosidase enzyme activity level in this liver and found that the enzyme level was elevated in the pack transplant group (Fig. 6E). We quantified the lyso-Gb3 level in the liver of the MESS transplantation group and the nontransplantation group, but there was no significant difference between them (Fig. 6F), suggesting that delivered enzymes were not enough to decrease the substrate of alpha-galactosidase.
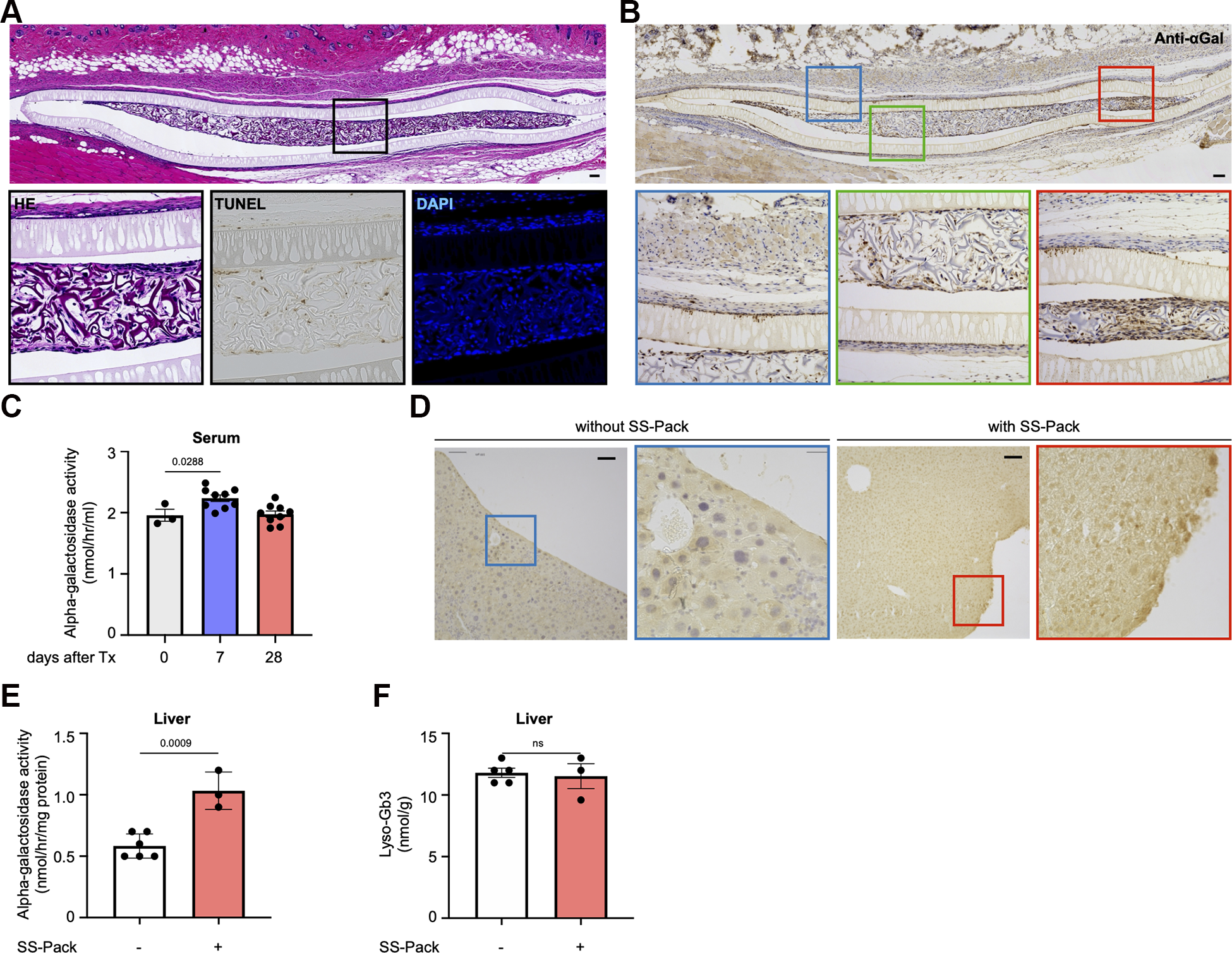
MESS transplantation of a genetically engineered cell-encapsulated immunoisolation membrane into the Fabry mouse model. (A) HE, TUNEL and DAPI staining of aGla gene-overexpressing immortalized MEFs (C57BL/6) encapsulated in pack transplanted in Fabry disease model mice (C57BL/6 background). The color of each square corresponds to the enlarged picture. Black bar indicates 100 µm. (B) Anti-aGLA antibody staining of aGla gene-overexpressing immortalized MEFs (C57BL/6) encapsulated in pack transplanted in Fabry disease model mice (C57BL/6 background). The color of each square corresponds to the enlarged picture. Black bar indicates 100 µm. (C) Alpha-galactosidase enzyme activity in serum after SS-immunoisolation membrane pack transplantation on each day. Tx indicates MESS transplantation. (D) Anti-aGLA antibody staining of the liver at 28 days after MESS transplantation. Black bar indicates 100 µm. (E) Alpha-galactosidase enzyme activity in the liver 28 days after MESS transplantation. (F) Lyso-Gb3 levels at 28 days after MESS transplantation.
Discussion
In this study, we created a novel unique therapeutic pack to treat Fabry disease, which is highly adjustable, exchangeable, and expandable in support of tissue engineering materials. Our system in this study can be improved with further genetic modifications to enclosed cells in macroencapsulation. The combination of macroencapsulation and scaffolded spheroids provides significant advantages over the methods alone. This methodology could lift cell therapy for LSDs closer to the clinical arena.
Tissue engineering holds the potential to generate functional tissues using cells, scaffolds, or both 30 . In the case of the combination of scaffolds and cells, scaffold matrices in tissue engineering play a role as structural support for cells, resulting in functionality and long-term survival. When a large volume of cells must be introduced, dispersed cells alone tend to aggregate, resulting in central necrosis of the aggregates and strengthened acidification of the encapsulated microenvironment 31 . For islet cells in the transplantation setting, the amount of insulin production per single cell is greater in 3-dimensional spheroids than in 2-dimensional culture 32 . The scaffold materials require biocompatibility and often biodegradability in the process where extracellular matrices to be produced by cells replace the intercellular space. To generate a new tissue to release lysosomal enzymes, biodegradability is dispensable, and maintaining a significant number of viable cells is strictly needed. A recombinant collagen peptide, μ-piece, provided long-term survival and functionality in the form of spheroids, called CellSaics 33,34 . In our previous study, CellSaics comprising μ-pieces and fibroblasts showed potential to release lysosomal enzymes, although the content was far from the therapeutic dose 20 . Alginate also offers an appropriate microenvironment for cells, such as islet cells, by separating host immune cells. Inflammatory foreign bodies respond to form pericapsular fibrotic overgrowths, which suppress the transport of oxygen and nutrients 35 . Chemical modification of alginate with triazolethiomorpholine dioxide mitigated foreign body responses, successfully resulting in long-term glycemic control 36 . Transforming growth factor-β-bound alginate induced immunotolerance with invasion of immature dendritic cells and regulatory T cells into the microcapsule 37 . Another concern of microencapsulation is the inability to retrieve the device due to its small size, in case one should happen to need to, such as an allergic reaction. Macroencapsulation offers an advantage to be able to be retrieved.
For a macroencapsulation device, a broad range of materials have been investigated to fulfill biocompatibility, immunoisolation, and oxygen/nutrient supply 24 . Inorganic materials, such as titania and silicon, take advantage of easily controlled physical characteristics, such as pore size and thickness 38 , but are prone to promote a fibrotic response around materials. Alternatively, polymers, such as polytetrafluoroethylene (PTFE) 39 and polycaprolactone (PCL) 40 , induce only limited fibrotic responses. The diameter of immune cells ranges from 6 to 10 μm, whereas structured proteins and organic metabolites range from 2 to 10 nm and from 0.5 nm to 1 nm in diameter, respectively 41 . Materials with submicrometer pore sizes could meet both immunoisolation and oxygen/nutrient supply requirements. PCL-derived thin films with nanosized pores for macroencapsulation devices showed stable functionality for 90 days in an allogeneic mouse model 40 . A membrane generated by microfabrication technology with nanochannels of 3.6 to 40 nm and microchannels of 20 to 60 µm also exhibited the immunoisolation and survival of encapsulated islets in a xenogeneic combination 42 . Another requirement of macroencapsulation is to secure the oxygen supply to encapsulated cells. Just after implanting the device, the vascular network does not exist surrounding it, leading to apoptosis 43 . The distance from the device membrane to most far cells can be no more than a few hundred μm 44 , which limits the design of the device. In the subcutaneous tissue, pre-vascularization of the transplant sites facilitated graft survival 45 , and the administration of growth factors accelerated the growth of the vasculature 46 . Other sites to be implanted could be the renal subcapsular space, intraperitoneal cavity, and omentum, which provide rich vascular growth, compared with the subcutaneous space. Given that retrievability is a priority, the subcutaneous space could be chosen with some ingenuity. An immunoisolation membrane called an EVAL membrane, which is a prototype of porous EVAL used in this study, are made of ethylene-vinyl alcohol that has a smooth surface and high hydrophilicity due to the presence of hydroxy groups in the side chains. The EVAL membrane was examined for macroencapsulation of islets in rat models and partially succeeded in preventing the formation of fibrotic reactions around the device 47 . The membrane had a drawback of inappropriate water permeability, which was 10 to 50 L/m2/h, and a porous EVAL membrane with 1,000 to 2,000 L/m2/h was developed. The membrane is clinically used as a dialysis membrane, taking advantage of its properties such as low adsorption of plasma proteins, low platelet activation, low generation of reactive oxygen species by active neutrophils, and low expression of inflammatory cytokines (IL-6) in monocytes 48 . Although the membranes are considerable for macroencapsulation devices, they have almost no oxygen permeability 49 . Furthermore, several kinds of porous immunoisolation membranes, one of which was used in this study, have been developed to enable various molecules to pass over it. The porous immunoisolation membrane used in this study possesses a maximum pore area of 0.5 μm2, fulfilling the submicron diameter required for macroencapsulation membranes. When rat pancreatic islets were encapsulated in a device using the same porous immunoisolation membrane as this study and transplanted into type I diabetic model mice, improvement of blood glucose levels and recovery of body weight were reported 50 .
A cross-correction of cells defective for lysosomal enzymes has been successfully achieved in some lysosomal diseases through either an in vivo or ex vivo gene therapy strategy 51 . Recently, a clinical trial on ex vivo gene therapy using autologous hematopoietic stem cells transduced with recombinant lentivirus carrying aGLA was conducted and published with promising results 19 . Although the number of patients was limited to 5, aGLA activity in plasma and leukocytes was sustained even after ceasing supportive enzyme replacement therapy in three patients. However, stored substance, Gb3, and the metabolite, Lyso-Gb3, in urine increased for two patients among three patients who stopped supportive enzyme administration. The remaining patient, who showed a decrease in these substances in both plasma and urine, exhibited antibody formation against aGLA in an early phase of transplantation, with a decrease in the later phase, suggesting a concern of adverse immunological reaction. Lysosomal diseases with neurological disturbances, such as mucoplysaccaridosis 52 and metachromatic leukodystrophy (MLD) 53 , have also been targeted by ex vivo gene therapy using HSCs because bone marrow-derived HSCs provide microglia that reside in the brain 54 . Although the cell product for MLD, which is generated by Orchard therapeutics, was approved by the European Medicines Agency (EMA) in 2020, the most concerning adverse event is the occurrence of anti-arylsulfatase-A antibodies. Another clinical trial for Fabry disease with autologous ex vivo gene therapy using HSCs, which was conducted by AVRO Bio, reported little clinical impacts for the formation of anti-aGLA antibody in four patients among 101 in a phase 1 trial and 2 patients among 111 in a phase 2 trial (https://www.avrobio.com). In addition, microencapsulation technology containing genetically modified cells that overexpress aGLA exhibited effectiveness with a reduction in Lyso-Gb3 in the heart and kidney, where ERT hardly exerts, to treat Fabry disease model mice (WORLDSymposium 2021). As a general concern of gene therapy, exogenous protein overexpression by gene transfer leads to excessive overload for the endoplasmic reticulum under ER stress, which could lead to acute inflammatory responses and activate innate immune responses 22 . For a safety net to the occurrence of the neutralizing antibody and inflammations, a strategy to be able to retrieve the grafted cells should be attractive.
In this study, we present a novel platform for ex vivo gene therapy using retrievable and exchangeable macroencapsulation in combination with scaffolds. Although simple overexpression of aGLA in donor cells could not achieve a significant reduction in the stored substrates, there is room to ameliorate enzyme release, such as modification of the M6P receptor. This macroencapsulation with scaffolded spheroids can extend to other types of lysosomal diseases.
Supplemental Material
Supplemental Material, sj-docx-1-cll-10.1177_09636897211060269 - Genetically Modified Cell Transplantation Through Macroencapsulated Spheroids with Scaffolds to Treat Fabry Disease
Supplemental Material, sj-docx-1-cll-10.1177_09636897211060269 for Genetically Modified Cell Transplantation Through Macroencapsulated Spheroids with Scaffolds to Treat Fabry Disease by Daisuke Kami, Yosuke Suzuki, Masashi Yamanami, Takahiro Tsukimura, Tadayasu Togawa, Hitoshi Sakuraba and Satoshi Gojo in Cell Transplantation
Footnotes
Abbreviations
Acknowledgments
Dr. Goro Kobayashi and Dr. Masako Kawagoe, who are employees of Kuraray Corporation, provided technical support related to the immunoisolation membrane.
Authors Contribution
S.G. designed the research and analyzed the data; D.K. performed all experiments; D.K. and S.G. wrote the paper; Y.S., M.Y., T.Tsu., and T.To. supportively performed the animal experiments; and H.S. reviewed and provided suggestions on the experiments and the manuscript.
Ethical Approval
This study was approved by our institutional review board.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: S.G. and D.K. received a collaborative research grant from Kuraray Corporation.
H.S. received research grants and personal fees from Sumitomo Dainippon Pharma Co., Ltd., and Sanofi Japan Co. outside of the submitted work. T.To. received a research grant from Sanofi Japan Co. outside of the submitted work.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding was provided by a collaborative research grant from Kuraray Corporation.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.