
Abstract
Traumatic brain injury (TBI) is a pervasive and damaging form of acquired brain injury (ABI). Acute, subacute, and chronic cell death processes, as a result of TBI, contribute to the disease progression and exacerbate outcomes. Extended neuroinflammation can worsen secondary degradation of brain function and structure. Mesenchymal stem cell transplantation has surfaced as a viable approach as a TBI therapeutic due to its immunomodulatory and regenerative features. This article examines the role of inflammation and cell death in ABI as well as the effectiveness of bone marrow-derived mesenchymal stem/stromal cell (BM-MSC) transplants as a treatment for TBI. Furthermore, we analyze new studies featuring transplanted BM-MSCs as a neurorestorative and anti-inflammatory therapy for TBI patients. Although clinical trials support BM-MSC transplants as a viable TBI treatment due to their promising regenerative characteristics, further investigation is imperative to uncover innovative brain repair pathways associated with cell-based therapy as stand-alone or as combination treatments.
Keywords
Introduction
Acquired brain injury (ABI) encompasses any injuries that disturb normal neuronal activity and is not classified as hereditary, degenerative, congenital, or caused by birth trauma 1 . ABI consists of stroke, traumatic brain injury (TBI), aneurysm, tumor, myocardial infarction, and many brain infections. ABI also includes alterations in metabolic activity, disruption to neuronal abilities, or changes in brain structure.Specifically, TBI is a prevalent and damaging form of ABI. A 2016 Global Burden of Disease Study found that there were 27.08 million new TBI cases worldwide with a prevalence of 2,349,017 cases in the United States 2 . Over 5 million Americans currently live with TBI-related disabilities 3 . In Europe, 2012 data revealed that TBI is responsible for approximately 2.1 million yearly hospital discharges as well as 37% of all deaths related to the injury 4 . Common characteristics of TBI consist of bleeding, bruising, and other forms of physical damage to the brain that may result in extended impairment or death. Cognitive features of TBI can include memory, concentration, attention, and thinking deficits, as well as behavioral changes such as mood swings or variations in sleep patterns 3 . The severity of TBI may be estimated by using the Glasgow Coma Scale (GCS), taking into account the duration of impaired/loss of consciousness, and investigating the mechanism of injury 5 . TBI is also associated with an increased frequency of developing neurodegenerative diseases including Alzheimer’s disease (AD) and Parkinson’s disease (PD), consequently increasing healthcare costs and chronic disability 6,7 . Although TBI is classified as acute cell death, subacute and chronic cell death are also associated with TBI given the presence of severe inflammatory responses. After TBI, the acute inflammatory response begins immediately, but unresolved damage can lead to subacute or chronic cell death during TBI progression 1 . Both the central nervous system (CNS) and the peripheral nervous system (PNS) contribute to TBI pathology, and cell-based regenerative medicines serve as potential therapeutic treatments for TBI. Modifications and the use of modern technologies have explored the applicability of bone marrow-derived mesenchymal stem/stromal cells (BM-MSCs) for the treatment of TBI 1 .
Acute Cell Death Classification of TBI
Considering the chronological sequence of the cell death cascade following a TBI, the initial insult resulting in primary cell death is recognized as the acute stage, while secondary cell death occurs progressively over the long term during the subacute and chronic stages 1 . The acute stage of TBI is characterized by sudden and severe cell death, also known as necrosis 1 . Acute cell death can be categorized as either focal or diffuse. Focal refers to acute cell death that occurs within a localized area of the brain, whereas diffuse denotes cell death arising throughout various regions of the brain, including areas that are isolated from the initial injured region of the brain 1 . Additionally, subacute and chronic cell death can spread to regions outside of the brain after the onset of TBI 1 . Specifically, remote secondary cell death has been observed to occur within the spleen, an organ that plays a key role in the inflammatory response 1 . This highlights the influence of peripheral factors on the processes underlying subacute and chronic cell death 3,8,9 . It is important to consider the severity of subacute and chronic cell death with increasing age. A younger brain exhibits more plasticity and may more effectively utilize endogenous repair mechanisms than an adult brain. Although TBI is classified as an acute brain injury, subacute and chronic cell death continues to develop over a drastic time period, and inflammation plays a major role in worsening these degenerative pathways 9 –12 . Given this information, targeting inflammation could point cell-based therapies in the right direction to reduce TBI-associated subacute and chronic cell death.
Inflammation, Subacute and Chronic Cell Death
The immune system’s capacity to propagate severe, inappropriate inflammatory responses in peripheral tissues is well established; however, recent evidence now details a similarly compelling role in CNS dysfunction 13 –16 . The CNS is immune privileged to a large extent, and it relies on specialized resident glial cells known as microglia to carry out immunoprotective functions. Typically, peripheral immune cells cannot penetrate the blood–brain barrier (BBB) to enter the cerebrospinal fluid (CSF); however, the acute inflammatory response following a TBI breaches the BBB, allowing for the infiltration of peripheral immune cells into the CSF 16 . In addition, the acute cell death induced by TBI stimulates proinflammatory microglial cells to mirror the activity of peripheral macrophages by migrating to the site of injury and exacerbating inflammation 15,16 . Furthermore, inflammatory cytokines released by microglial cells collaterally act on the BBB to increase its permeability enough to allow circulating leukocytes to migrate from the peripheral circulation into the CNS 17,18 . These leukocytes exhibit a remarkable aptitude toward modulating the glial cells by promoting continuous inflammation that impairs neuronal recovery and exacerbates Wallerian degeneration of axons 9,19 . Therefore, targeting the immune pathway to ablate the inflammatory response postinsult may be a potent approach to reduce subacute and chronic cell death.
Acute Inflammation
The acute inflammatory response begins immediately after TBI. Following initial insult, focal ischemia signals microglia to promote expression of the surface protein cluster of differentiation 14 (CD14) 19 . CD14 is a pattern recognition receptor that is typically found on circulating monocytes, but following TBI, significantly elevated numbers of CD14+ cells—both activated microglia and peripheral monocytes invading the injury site—have been observed in the perivascular spaces and brain parenchyma, indicating CD14’s role in the neuroimmune response to CNS injury and suggesting that microglia fulfill the role of acute inflammatory mediators within the CNS 19,20 . CD14 enhances microglia sensitivity to lipopolysaccharides (LPS), which allows microglia to recruit proinflammatory cytokines even more effectively than peripheral macrophages 21 . Although CD14 also exhibits a neuroprotective role against injury by suppressing LPS-induced nitric oxide generation, alleviating glial neurotoxicity, and inhibiting microglial and astrocyte cell death, the increased expression of this receptor is indicative of TBI-induced acute inflammation, as CD14 generates a heightened inflammatory state in the CNS via microglia recruitment 21,22 . In addition to expressing this innate immune system receptor, M1-activated microglia secrete the proinflammatory cytokines tumor necrosis factor-alpha (TNF-α) and interleukin 1 beta (IL-1β) into the CSF and systemic circulation while neurons simultaneously stimulate interleukin 6 (IL-6) expression 17 . Importantly, although M1 microglia are proinflammatory, the M2 variant secretes neuroprotective cytokines including transforming growth factor-beta (TGF-β) and interleukin 10 (IL-10) 16,23 . Therapeutic strategies that promote differentiation toward M2 may be valuable; however, the dynamic milieu of cytokines, growth factors, and cell signals makes it difficult to maintain discrete phenotypes in vivo 24 . Indeed, the postinjury microenvironment is replete with a diverse range of proinflammatory and neuroprotective factors.
As the acute inflammatory response begins to wane, the pathological cascade progresses toward the subacute phase. During this time, inflammation progresses through the action of proinflammatory cytokines and the secretion of cellular adhesion molecules (CAMs) and matrix metalloproteinases (MMPs) by damaged neurons, microglia, and immune cells 17,24,25 . Circulating leukocytes bind to CAMs to isolate themselves to the site of injury and then extravasate from the vasculature into the tissues. MMPs facilitate this process of leukocyte escape by permeabilizing the BBB, thereby enhancing the entry of inflammatory cells into the cerebral space 25 . This combination of increased permeability, augmented leukocyte localization, and profound inflammation ultimately fosters edema, swelling, and neuronal damage 9,26 –28 . The continued action of activated microglia, CAM and MMP secretion, and numerous key cytokines eventually leads to the progression into the chronic phase of inflammation.
Although TBI causes acute cell death from the direct physical damage, the mechanistic response associated with the subacute and chronic cell death after the initial impact has been a subject of interest for understanding the pathology and treatments of TBI-induced secondary cell death 12 . Briefly, an insult is quickly followed by a largely insufficient transient neuroprotective reaction mediated by a small subset of microglia that differentiate into the neurotrophic M2 phenotype 16 . These microglia promote neurogenesis and cellular regeneration in key brain regions such as the dentate gyrus (DG) of the hippocampus 15 . However, the protection afforded by this short-lived response is countered by the postinjury chronic inflammatory state. At the site of injury, most microglia differentiate into the M1 subtype acutely, and this proinflammatory subtype may persist for up to 20 years postinjury 12,15 . Microglia localize directly to the site of injury and to the surrounding tissues as well 11 .
Microglial Polarization
As discussed previously, microglia exist as two different phenotypes: M1 (proinflammatory) and M2 (immunosuppressive/neuroprotective) 29 . The polarization of microglia is dependent on a variety of stimuli. M1 microglia are associated with the secretion of proinflammatory cytokines (i.e. TNF-α and IL-1β) and the generation of oxidative factors (i.e. nitric oxide and reactive oxygen species) 29 . Moreover, M1 microglia serve as the initial protective mechanism against injury but simultaneously generate a heightened inflammatory state. On the other hand, M2 microglia initiate anti-inflammation, damaged tissue rehabilitation, and the restoration of extracellular matrix (ECM) 29 . Imbalance of M1/M2 polarization is characteristic of neurodegenerative diseases, such as AD and PD. In TBI, M1 phenotype microglia become prevalent early to sequester cellular and molecular detritus 30 . This early M1 microglial activation is vital for the repair of disrupted brain homeostasis 30 . This activation also creates a toxic environment due to the secretion of inflammatory cytokines and oxidative agents 30 , and if microglia remain activated for too long, inflammation and tissue injury can be exacerbated, leading to neurodegeneration 30 . The imbalance of microglial polarization has risen as a potential therapeutic target in TBI.
Subacute and Chronic Inflammation
Unresolved acute inflammation progresses to the subacute and chronic phases which are characterized by severe TBI progression. Severe TBI can result in a coma or a minimally conscious state, followed by a post-traumatic confusion and post-traumatic amnesia 31 . The dynamic relationship between peripheral lymphocytes and CNS microglia determines patient outcomes, with death resulting from an inappropriate balance of these systems 9,10,16 . This is observed in patients as some present with cognitive decline for days to years following TBI 9 . Furthermore, microglia have been shown to remain active for up to 1 year in animal models 9 . The communication between the central and peripheral cells occurs through the weakened BBB, which allows the systemic leukocytes and proteins to enter the CNS 18 . The subsequent injury from the chronic pathological presence of these compounds in the cerebral tissues exacerbates intracranial pressure and is tightly correlated with cognitive decline and neurodegeneration 9,18,28 . The subacute and chronic cell death from TBI is reminiscent of that seen in ischemic stroke, which triggers an anti-neuron autoimmune response that may either augment or diminish the neuroinflammation 11,32 .
The peripheral leukocytes’ migration into the cerebral tissue through the disrupted BBB to promote inflammation and activate microglia is key to the pathology of TBI. Once in the brain parenchyma, the neuroinflammation induced by these cells creates a deleterious environment unfavorable to cellular survival. This likely underlies the poor rates of stem cell graft transplant survival in TBI patients and the heightened hippocampal degeneration in the ipsilateral subventricular zone (SVZ) and subgranular zone (SGZ) 11,33 . It may also explain the rapidly emerging link between TBI and the amyloid-beta (Aβ) plaques and neurofibrillary tangles distinctly observed in the brains of AD patients 34 . Although AD is typically thought of as an age-related disease, the presence of Aβ plaques in the brains of a broad age range of TBI patients, including children, suggests a potential link to physical injury 15 . Aβ42 aggregation impairs microglial cell’s ability to carry out phagocytosis 15 . Importantly, TBI is associated with a number of other major neurological issues, some of which may require several months to manifest 35 . Postmortem analysis of TBI patient’s brains within 2 months of injury demonstrated the presence of proteins associated with neurodegenerative diseases, such as the PD-specific α-synuclein. Remarkably, the dopaminergic neuronal cell loss in PD potentiates a microglial activation similar to that observed in TBI; it is defined by proinflammatory cytokine release in the cerebral tissues and a reactive gliosis 3 . In PD, damaged neurons secrete α-synuclein, which aggregates into protofibrils at the presynaptic cleft 36,37 . These harmful accumulations enter the CSF to produce harmful consequences, as identified in the brains of infants and children. This pathological accumulation of α-synuclein in cell bodies or axons may represent a critical relationship between PD, AD, and TBI. Models of spinal cord injury support the finding that microglia are the primary CNS cells that respond to injury, and they remain activated for several months after insult 38 .
Central and Peripheral Sources of Inflammation
After a neurological insult like TBI, both the CNS and PNS play a substantial role in the inflammatory cascade that causes further damage to the neural tissue 9,39,40 . The central inflammatory response is identified by microglia and other resident brain immune cells, such as dendritic cells, CNS border-associated macrophages, natural killer cells, and mast cells 41,42 . The peripheral inflammatory response incorporates more of a systemic response, recruiting immune cells from important organs such as the spleen and thymus 9 . In order to uncover the subacute and chronic cell death cascade that exacerbates the initial TBI, it is critical to investigate the central and peripheral organs involved in neuroinflammation and how those organs contribute to the overall pathology of TBI. It is important to recognize the role that the ligand-receptor pair CCL20-CCR6 has in sending chemical stimuli that recruit immune cells such as dendritic cells and effector/memory T and B cells during an inflammatory event such as TBI 9 .
CCL20 acts as a cytokine for CCR6-expressing leukocytes. In an animal model used to replicate neuroinflammation, experimental autoimmune encephalomyelitis (EAE) model, CCL20 acts as a ligand for the CCR6 receptor, contributing to the signaling of immune cells and Th17 or Th1 cluster of differentiation 4 (CD4+) cells that further express proinflammatory cytokines that are observed in chronic brain inflammation 43,44 . As more CCL20 is present in the choroid plexus, CCR6+ T cells are allowed to infiltrate the CNS in the EAE model and therefore increase T cells occupancy in the brain parenchyma. Furthermore, increased expression of proinflammatory cytokines IL-6 and interleukin 17 (IL-17) upregulate CCL20 translation 43,44 .
In a lateral fluid percussion model of TBI, CCL20 expression is increased in the spleen and thymus 24 h after insult and the cortex and hippocampus 48 h after insult, suggesting a potential mechanism of action that is fundamental to the role of the periphery in neuroinflammation 39,40,43 . It was observed that CCL20 was expressed in the spleen and thymus after TBI before presenting in the brain 39 . Furthermore, after splenectomy, there was reduced expression of CCL20 in the periphery, suggesting that the peripheral immune response may serve as a precursor to CCL20 expression in the CNS, resulting in increased neuronal damage 39 . In addition, other studies suggest that the liver may also increase neuroinflammation after TBI. It was found that deficient Kupffer cells originating from the liver reduced ED-1-positive macrophage and neutrophil migrate into an IL-17-injected brain 44 . Kupffer cells are the most prevalent tissue-resident macrophage and are located in the liver sinusoids. These cells aid in the removal of pathogens and maintaining tissue homeostasis 45,46 . The peripheral immune response, notably the spleen, works together with the central inflammatory response induced by microglia and inflammatory cytokines. All in all, after TBI, the peripheral and central inflammatory mechanisms contribute to a chronic state of neuroinflammation that results in further damage to neurons and delayed brain repair.
Cell-Based Therapy for TBI
Given that TBI pathology manifests both centrally and peripherally, it may be efficacious to administer cell-based regenerative medicines both intracerebrally and systemically, either by intravenous or intra-arterial injection 47 –49 . In the aged brain, combination therapies of cell transplants and other treatments, such as hypothermia 50,51 and electrical stimulation 52 –54 , may be needed in order to promote brain repair. Whereas, in younger populations, cell-based therapy may be all that is needed to facilitate neuronal regeneration 3 . In addition, cell transplants may dampen both short-term and long-term inflammation (Fig. 1) 3,24 –26 . Immediate administration of cells following TBI seeks to offer neuroprotection by preventing inflammation, apoptosis, mitochondrial dysfunction, and oxidative stress 55 . Meanwhile, long-term administration of cells serves to promote synaptogenesis, neurogenesis, angiogenesis, vasculogenesis, and overall brain repair 55 . It is apparent that increased inflammation is a common factor across most populations and injury sites, and a cell therapy that targets this inflammation can ameliorate and even decelerate the disease progression of TBI by reducing subacute and chronic cell death 55 .
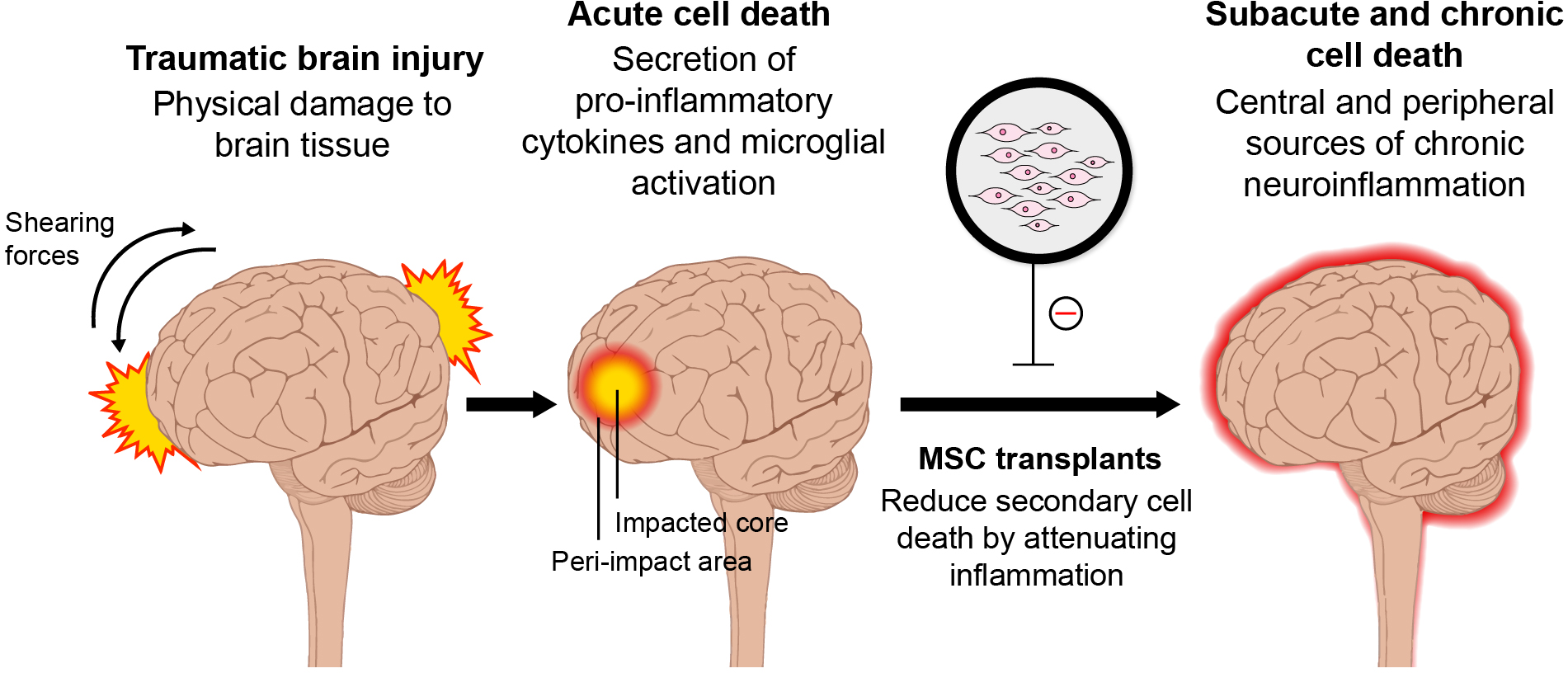
Schematic diagram of TBI-associated secondary cell death and MSC therapy. Transplantation of MSCs may sequester the secondary cell death of TBI by reducing neuroinflammation acutely, subacutely, and chronically. MSC, mesenchymal stem cell; TBI, traumatic brain injury.
A potential approach to cell-based TBI therapy consists of stimulation of endogenous stem cells 55 . Stimulation of endogenous stem cells shows great therapeutic promise as the mature adult brain has been shown to possess regenerative capabilities in the SVZ and the DG of the hippocampus 56 . Adult stem cells possess the capacity to proliferate and differentiate to renew injured and senescent cells 56 . Moreover, TBI has been shown to induce endogenous repair mechanisms resulting in stem cell proliferation around the SVZ and DG in murine models 57 . Targeting endogenous stem cell populations may provide an effective therapeutic option to treat TBI 55 .
Another promising avenue of cell-based therapy is the transplantation of exogenous stem cells 55 . A wide variety of transplanted exogenous stem cells have been investigated to overcome the limited regenerative capabilities of endogenous brain repair within TBI 55 . Exogenous stem cell transplantation has also demonstrated regenerative capabilities in other neurodegenerative disorders such as stroke 55 . Their capacity to differentiate and home to the site of injury could provide favorable effects on the injured brain by replacing dysfunctional and senescent neurons. Aside from renewing injured cells, exogenous stem cells may enhance functional improvements via the secretion of many anti-inflammatory molecules, chemokines, cytokines, and growth factors. These molecules can significantly dampen the harsh environment of the inflammation-ridden brain and promote endogenous repair mechanisms 55 .
Targeting Inflammation with Cell-based Therapy in TBI
Targeting inflammatory mechanisms in TBI may provide insight on subacute and chronic cell death progression as well as potent treatments. Uncontrolled inflammatory activity in TBI, originating from peripheral and central organs, has been correlated with functional recovery deficits and post-TBI cell deaths. Peripheral organs, specifically the spleen, may serve as potential target tissues for analyzing subacute and chronic cell death mechanisms due to their significant contribution to systemic inflammatory activity. To further understand the pathologies of neuroinflammation and develop anti-inflammatory strategies for TBI, recognizing the importance of central and peripheral systems in TBI progression and tissue damage exacerbation is vital.
Bone Marrow-Derived Mesenchymal Stem Cells for TBI
Although the success of stem cell transplantation for TBI-induced neuroinflammation is dependent on translational factors from lab to clinic 58 , the type of stem cell being administered may influence transplantation efficacy the most. Some translational factors include determining the optimal route, dosage, and timing of stem cell treatment 58 . In addition, the safety index of the stem cells must be fully analyzed, as some can be tumorigenic 58 . Ideally, the stem cells should exhibit robust stemness/pluripotency, which is the ability to differentiate into any of the three germ layers 59 . Numerous types of stem cells have been examined to determine their efficacy as cell donors in TBI cell transplantation. For example, embryonic and fetal stem cells possess desired pluripotent characteristics. However, the method of harvest and potential adverse effects, such as the development of tumors, raise ethical and safety concerns in regard to their clinical use, shifting the target to other stem cells, namely adult tissue-derived cells. BM-MSCs exhibit therapeutic effects on the brain similar to that of embryonic and fetal stem cells without notable ethical or safety considerations 58 in addition to its availability and well-supported history, making it a favorable tool for stem cell treatments.
Multiple neurological disease models have highlighted the restorative effects of BM-MSCs in functional recovery 60 –66 . Animal models demonstrated structural brain repairment and improvement in motor and cognitive functions after BM-MSC administration. Specifically, reduced proinflammatory cytokine expression levels, including IL-6, interleukin 1 alpha (IL-1α), and interferon-gamma (IFN-γ), were observed after BM-MSC transplantation via intraventricular infusion 67 . Growth factors released by BM-MSCs may be responsible for the observed therapeutic activity, including neurotrophic exosomes, neurogenesis, angiogenesis, and direct cell replacement 58 . However, specific mechanisms and pathologies have not been discovered. Some studies offered models, such as reductionist cell implantation, to explain the mysterious therapeutic mechanisms of cell implantation. Unfortunately, proposed models were determined to be over simplistic. Little evidence supporting the mechanism of MSC and study designs presenting limitations hindered patient recruitment in clinical trials and inferior brain examinations. Grafted cells were proposed to inherit growth factors that harmoniously reduce inflammation, oxidative stress, and apoptosis in addition to other bystander mechanisms to restore the brain. Bystander mechanisms refer to the ability in which grafted cells bring about both functional and structural restorative benefits and their role in immunomodulation, neurogenesis, brain plasticity, and secretion of neuroprotective factors 68 .
Given that post-TBI inflammation is responsible for subacute and chronic cell death, which further induces cognitive dysfunctions, BM-MSCs are potent treatment options for TBI due to their anti-inflammatory and immunosuppressive abilities. BM-MSC administration via intravenous infusion was seen to lower microglia and immune cells activity at damaged sites in TBI rats 69 . Furthermore, BM-MSC treatment may downregulate or inhibit proinflammatory cytokines while also inducing anti-inflammatory cytokines possibly by suppressing microglial activation 70 . Restorative effects of BM-MSC treatment in TBI rats were also observed by increased neuroprotective properties, improved functional recovery, and neural regeneration 71 –74 . In addition to its therapeutic properties, BM-MSC may have enhanced efficacy in treating TBI if the stem cells were genetically modified. Homing capabilities of BM-MSCs to TBI lesion sites were further improved when the stem cells expressed higher levels of fibroblast growth factor 21 (FGF21) 70 . Other TBI rat models highlighted enhanced autophagy, mitophagy, and immunomodulation activity in BM-MSC with overexpressed anti-inflammatory cytokine IL-10, significantly enhancing neuroprotection and functional recovery 75,76 .
Coupling cell treatment and other neurological treatments, such as biomaterial, pharmaceutical interventions, and additional cell transplantations, may also significantly improve the cognitive and motor functions of patients undergoing cell therapy 58 . In stroke models, anti-inflammatory activity was evident after coupling BM-MSC with other interventions, such as peroxisome proliferator-activated receptor gamma agonist pioglitazone and regulatory T cells 77,78 . Combining BM-MSC with other therapeutic methods may introduce enhanced restoration of cognitive function in stroke and TBI-induced neuroinflammation models 12,79 –86 . However, further investigations must be conducted to verify if similar potent effects result when applied to TBI models.
Clinical Perspective on MSC Therapy for TBI
The efficacy and viability of BM-MSCs in the clinical setting still face significant questions and challenges, despite preclinical evidence supporting the feasibility of BM-MSCs in use as cell-based therapy. Transplanted BM-MSCs have demonstrated promising results in animal models. They provide a wide array of functional mechanisms, ranging from endogenous stem cell mobilization, secretion of trophic factors, and to encourage recovery after CNS insults 58 . Moving to the clinical setting proves difficult due to the painful procedure of collecting bone marrow samples. By analyzing the recent data from clinical trials in human subjects, these discrepancies may be resolved for a better understanding of BM-MSC transplantation and the procedure’s utility as a treatment for TBI in the future.
The interactions between grafted or endogenous cells with immune cells have broad implications for regenerative medicine. Both the adaptive (B and T cells) and innate (macrophages, monocytes, microglia) immune systems regulate autonomous mechanisms for cell and non-cell regenerative responses, with postinjury immune responses triggering either increased or reduced inflammation. The circulating population of immune cells infiltrating the graft can also influence grafted cells to regulate the immune response 87 –90 . Major histocompatibility complex (MHC) II expressing macrophages that are cocultured with adipose-tissue derived MSCs demonstrate upregulated collagen settlement via MHC II signaling. Proliferation and expression of MMPI (incites migration of cells), PLOD2 (maintains intermolecular crosslinks), and PTGS2 (regulates the proinflammatory response) are also shown to be accelerated 89 . This evidence supports the role of MSCs in modifying the innate immune response’s role to promote healing; however, there is also evidence that the adaptive immune system hinders the anti-inflammatory and therapeutic response from grafted cells. One method is T cells and NK cells may target stem cells such as MSCs due to MHC I expression, triggering immune-mediated cytolysis 87 . Despite this, MSCs may evade this immune regulation; a study provided evidence that MSCs can physically hinder T cells from contact with antigen-presenting cells which can explain why both naive and memory T-cell response is inhibited in a dose-dependent fashion when cultured together 87 . The interactions between immune cells and stem cell grafts require further investigation into the exact cellular processes, but current evidence has both positive and negative implications for the potential usage of MSCs in TBI.
In a recent clinical trial, autologous BM-MSC transplantation was supported to be safe and effective in a 2013 trial. Ninety-seven patients were administered BM-MSCs via lumbar puncture, with 38 (39.2%) of the patients demonstrating improved neurological function following transplantation 91 . In addition, 27 out of 73 (37.0%) patients presenting with motor disorders showed improvement in motor function, and 11 of 24 patients in a persistent vegetative state showed post-therapeutic improvements in consciousness. While the results do not show BM-MSC transplantation has efficacy in every patient, the study noted variable outcomes based on factors such as patient age and the post-injury administrative window, with a significant correlation of earlier administration and younger patient age with improvement 91 . Another clinical study using BM-MNCs (mononuclear cells) in TBI indicated both safety in bone marrow harvest and an infusion, which is promising for future trials identifying efficacy. Twenty-five patients received intravenous BM-MNC infusions in a dose-escalation trial (6, 9, 12 × 106 cells/kg body weight), without severe adverse effects 92 . Furthermore, paralleling pre-clinical evidence from animal models, the treatment also correlated with a downregulation of inflammatory cytokines IL-1β and IFN-γ. For patients with TBI, the feasibility and safety of performing bone marrow transplantation are supported by these findings 92 .
Updated Preclinical and Clinical Studies of the Utilization of MSC Therapy on TBI
Recent studies have further explored the use of MSCs as a therapy to treat TBI. Researchers have also uncovered mechanisms to enhance and maximize the efficacy of MSCs in vivo. The number of recently completed clinical trials that have utilized MSC transplantation is still lacking; however, there are many ongoing and recruiting studies. Updated preclinical evidence warrants the further investigation of MSCs as a potent therapeutic against TBI, and more clinical trials should be conducted. Modification and coupling of MSCs has also been an area of interest and demonstrated beneficial results in a preclinical setting.
MSCs were modified to upregulate and overexpress FGF21. FGF21 has shown to augment angiogenesis, elicit neuroprotective effects, and induce remyelination. The FGF21-enhanced MSCs were administered via intracerebroventricular injection 24 h after TBI in mice. Three to 4 weeks after treatment, the spatial memory deficits, lessened hippocampal neurogenesis, and abnormal dendritic morphology associated with TBI significantly improved 93 . Human umbilical cord-derived mesenchymal stem cells (hUC-MSCs) have shown positive results in improving TBI-induced deficits. hUC-MSCs efficacy is impeded by the severe environment of the injured brain. The MG53 protein was investigated in combination with hUC-MSCs to elucidate whether it could facilitate the improved function of the stem cells and help attenuate the oxidative stress that hinders the maximum therapeutic effectiveness of MSCs. Data indicated that MG53 preserved hUC-MSC proliferation and migration by attenuating hydrogen peroxide associated-oxidative damage. Moreover, coupling MG53 and hUC-MSCs in rats relieved brain edema and improved deficits. Apoptosis was observed at a lower rate, and PI3K/Akt-GSK3β signaling was increased. Further exploration of stem cell coupled agents is encouraged to maximize dosage and route of administration to enhance MSC survival and function 94 . MSC therapy to target TBI is often impeded by host immune responses, specifically cytotoxic cluster of differentiation 8 (CD8+) T cells, which can interrupt therapeutic efficacy. To combat this, an agarose hydrogel was created to induce apoptosis of cytotoxic CD8+ T cells via secretion of Fas ligand. This treatment mechanism was investigated in tangent with MSC transplantation. When applied in the area of MSC transplantation, a localized decrease in cytotoxic CD8+ T cells was observed. Furthermore, data indicated augmented levels of neurotrophic factors which consequently improve TBI-associated deficits. Further investigation is warranted on the efficacy of immunosuppressive hydrogels to facilitate stem cell transplantation for neurodegenerative disorders 95 .
MSC-Derived Exosomes
Many preclinical investigations suggest that MSC-derived exosomes provide beneficial therapeutic potential 96 . The efficacy of human adipose mesenchymal stem cell-derived exosomes (hADSC-ex) was investigated in a TBI rat model. Data indicated that hADSC-ex attenuated the inflammation associated with TBI via inhibiting microglia and macrophages during injury 96 . By mitigating inflammation, hADSC-ex permitted reduced neuronal apoptosis and enhanced neurogenesis 96 . Furthermore, tested in a swine TBI model, recipients of exosomes displayed decreased lesion size, swelling, and intracranial pressure 97 . Data revealed decreased concentrations of acidic protein levels and augmented concentrations of zonula occludens 1, claudin-5, and laminin. MSC-derived exosomes have the capacity to relieve swelling and fortify BBB integrity. These findings further contribute to MSC-derived exosomes’ therapeutic potential and confirm that MSCs retain their capabilities as a potent treatment within a larger animal model 97 .
MSC-Derived Secretome
Another alternative to MSC transplantation is the administration of MSC-derived secretome. Umbilical cord mesenchymal stem cells were pretreated with TBI brain tissue to stimulate the MSC secretome. The traumatic injury-preconditioned secretome delivered increased efficacy in enhancing maturation, differentiation, and migration in the DG when compared to non-preconditioned secretome treatment. Taken together, these beneficial effects could contribute to improvements in cognitive recovery. Secretome therapy proves a beneficial avenue to treat TBI and may be a safer alternative than stem cell transplantation 98 . Adipose-derived mesenchymal stem cell secretome (ASC-ST) collected under hypoxic conditions was injected into TBI rats. Inflammation and vasogenic edema were attenuated by ASC-ST. Furthermore, ASC-ST increased M2 and decreased M1 microglia phenotypes which resulted in a decrease in neuronal apoptosis and nerve fiber damage. Secretome treatment augmented concentrations of TGF-β and tumor necrosis factor-stimulated gene 6 protein while decreasing IL-6 and TNF-α levels. By regulating TBI-induced secondary inflammation, ASC-ST proves very beneficial in treating TBI and other CNS diseases 99 .
Clinical Studies
A 2017 clinical trial provided an interesting correlation between hyperbaric oxygen treatment (HBOT) and mobilization of stem cells 100 . Twenty-eight subjects who suffered TBI during military deployment were analyzed for relative abundance of stem cells in the peripheral blood post-HBOT. Fluorescence-activated cell sorting analysis indicated that HBOT treatment did promote stem cell mobilization and consequently boosted cognition 100 . These effects of HBOT also served to relieve post-traumatic stress 100 . This mechanism should be further explored to possibly couple with MSC transplantation 100 . Although there has been a lack of recently published clinical trials 101 , there are many ongoing and recruiting studies 102 . An active trial has recruited 300 male and female participants who present with CNS damage, including TBI-induced injury. The goal of this study is to test the therapeutic efficacy of bone (BM-MSCs when delivered intranasally or intravenously). Daily activities will be evaluated over the course of a year, and neurological function will be assessed. This study is ongoing, and results have not yet been published 102 . A phase I ongoing trial is currently utilizing adipose tissue derived-mesenchymal stem cells (AD-MSCs) and treating participants 3 times for 6 weeks post-TBI 103 . They plan to analyze inflammatory cytokines as well as neuropsychology, cognition, and neurological function of patients after treatment. They aim to determine the safety of AD-MSC infusion as well as the influence treatment has on the structural integrity of gray and white matter in the corpus callosum 103 . Many studies are also investigating the therapeutic potential of bone marrow mononuclear cells (BMMNCs) within TBI 104 . The aim of one phase I completed trial was to determine the efficacy, safety, and feasibility of BMMNCs. Results indicated no clinically significant side events were induced by transplantation and showed BMMNC’s were a practical and safe treatment 104 . TNF-α concentrations were decreased in patients 104 , and this was also recently observed in rats upon MSC transplantation in TBI 99 . An active phase II trial also utilizing BMMNC’s and aims to investigate the macrostructural and microstructural properties of gray matter, white matter, and corpus callosum integrity. Results have not yet been published 105 .
Pharmacological Treatments for TBI
In contrast to cell-based therapies, the use of pharmacological treatments for TBI also demonstrates their efficacy in the laboratory. Cyclooxygenase (COX) inhibition reduces some of the harmful effects associated with brain injury 106 . Carprofen, an anti-inflammatory drug, improves TBI outcomes by decreasing the lesion size and increasing neurogenesis in rodents 106 . Additionally, Rolipram displays neuroprotective effects, improves synaptic plasticity, and improves cognitive performance in a mouse model of TBI 107,108 . As these pharmacological treatments have been tested in preclinical TBI models and have been shown to be neuroprotective, they remain to experimental therapeutics and await their translation into clinical trials for TBI patients 109 . Combining cell-based treatments with such drugs may enhance their efficacy and provide viable TBI treatment options.
Conclusion
Due to the substantial health and economic strain TBIs pose around the world, along with inadequate treatment options call for an advanced therapeutic approach to mitigate disease progression 110 –113 . Resulting acute cell death by TBI causes brain damage that can be categorized as either diffuse or focal. Secondary neurodegeneration is notably influenced by peripheral sources of immune cells and the age of the patient during both the acute and chronic phases after TBI 114 –118 . Neuroinflammation that occurs following TBI closely resembles subacute and chronic neural cell loss progression. Delineating the relationship between the brain, a central source, and the spleen, a peripheral source, is essential in understanding the mechanisms causing neuroinflammation. One novel regenerative biologic therapy that has emerged is MSC transplantation, which targets the neuroinflammation caused by TBI. Enhancements within the cell therapy tactic are necessary to help transplantation strategies accommodate varying brain inflammatory responses. In regard to focal brain injury, the localized intracerebral delivery of MSCs may be a more suitable strategy to directly target the inflamed area. With diffuse brain damage, systemic delivery of MSCs and targeting the spleen could help hinder both central and peripheral sources of inflammation. Although stand-alone MSC transplantation may be adequate enough to initiate neuroprotection and rejuvenation in younger brains, combination therapies could be essential to meet the gold standards of treatment. In addition, recurrent supplementation of MSC infusions throughout the chronic phase following TBI may be needed after the initial MSC injection that is given during the acute phase. Through accumulating preclinical evidence, BM-MSCs have shown the potential to model a propitious cell therapy option due to their anti-inflammatory and immunomodulatory properties. It is suggested to incorporate blinding and sample size calculations, randomization, heterogenous animal genders, at least two different animal models, comorbid animal strains, and investigation of appropriate dose-response relationships. Adhering to these STEPS guidelines will likely produce a safe and effective design framework for the clinical application of MSCs. Stem Cell Therapies as an Emerging Paradigm in Stroke (STEPS) provide guidance for preclinical testing including cell delivery approaches, such as cell dosing, timing, and route, as well as devices to assist cell delivery, and considerations for clinically relevant animal and human testing. These guidelines have been implemented to facilitate the successful translation of safe and effective cellular therapies from preclinical studies to clinical trials 119 . Use of MSCs with TBI represents a promising therapy technique for future therapeutic ventures that are aimed at treating TBI.
Currently, TBIs possess limited treatment options, especially TBI pathologies caused by acute, subacute, and chronic cell death. BM-MSCs have the potential to fill this treatment gap due to their initial neuroprotective effects and their long-term neuroregenerative and immunomodulatory effects. Successful translation of MSC transplants from the lab to the patient’s bedside could help diminish the health and economic burden caused by TBI. There are currently only a small number of clinical trials that use BM-MSCs for the treatment of TBI despite their positive preclinical results, relative accessibility, and well-established safety record in their use for other disorders. There is a limited capacity for cross-examination of BM-MSC trials due to the varying research designs and dissimilarities between research outcomes. This review emphasizes the necessity for more clinical trials, especially those who follow the STEPS guidelines. These findings stress the importance of addressing the issue of scaling up MSCs since it remains a rate-limiting step in some treatments. This review also brings attention to the interest surrounding MSC transplant administration. This includes the possibility of administration that is in concert with the adaptive and innate immune system of the patient, as well as the characteristics of the patient, injury specifics, dosage, number of doses, and timing. The use of MSC transplants as a therapeutic for TBI has grown exponentially over the past decade and is likely to continue to grow. Upon the discovery of viable approaches that improve cell proliferation, significant scientific milestones may be reached and MSC transplant therapy could become the standard of care for the treatment of TBI. MSC therapy represents a rich area for future research. The desired outcome of this research is the development of a successful and safe cell therapy that helps to rid the subacute and chronic cell death inflammatory response associated with TBI.
Footnotes
Acknowledgments
The authors thank the entire staff of the Borlongan Neural Transplantation Laboratory for critical discussion of this manuscript.
Author Contributions
All authors contributed to the conception, literature analysis, preparation and writing of original draft, and reviewed and edited the manuscript. CVB performed the supervision, project administration and acquisition of funds. All authors have read and agreed to the published version of the manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: CVB is funded by National Institutes of Health (NIH) R01NS090962, NIH R01NS102395, and NIH R21NS109575.