
Abstract
Human skin contains keratinocytes in the epidermis. Such cells share their ectodermal origin with the central nervous system (CNS). Recent studies have demonstrated that terminally differentiated somatic cells can adopt a pluripotent state, or can directly convert its phenotype to neurons, after ectopic expression of transcription factors. In this article we tested the hypothesis that human keratinocytes can adopt neural fates after culturing them in suspension with a neural medium. Initially, keratinocytes expressed Keratins and Vimentin. After neural induction, transcriptional upregulation of NESTIN, SOX2, VIMENTIN, SOX1, and MUSASHI1 was observed, concomitant with significant increases in NESTIN detected by immunostaining. However, in vitro differentiation did not yield the expression of neuronal or astrocytic markers. We tested the differentiation potential of control and neural-induced keratinocytes by grafting them in the developing CNS of rats, through ultrasound-guided injection. For this purpose, keratinocytes were transduced with lentivirus that contained the coding sequence of green fluorescent protein. Cell sorting was employed to select cells with high fluorescence. Unexpectedly, 4 days after grafting these cells in the ventricles, both control and neural-induced cells expressed green fluorescent protein together with the neuronal proteins βIII-Tubulin and Microtubule-Associated Protein 2. These results support the notion that in vivo environment provides appropriate signals to evaluate the neuronal differentiation potential of keratinocytes or other non-neural cell populations.
Introduction
Neural stem cells (NSCs) that differentiate to neurons, astrocytes, and oligodendrocytes can be obtained from different sources, including human primary fetal tissue 1 , postmortem neonatal tissue 2 , or adult neural tissue 3 –5 . Alternatively, neural induction of human embryonic stem cells (hESCs) or induced pluripotent stem cells (iPSCs) can produce NSCs. These sources have been used in different animal models to restore acute damage or to treat diseases such as Parkinson, Huntington, or Alzheimer 6 . Unfortunately, obtaining cells from human embryos or postmortem individuals is challenging due to their limited availability. On the other hand, it is well known that using ESCs and iPSCs comes with the risk to generate teratomas if pluripotent cells do not undergo differentiation.
A possibility to obtain NSCs is to generate neural cells by plasticity events from alternative cellular sources. Neural precursor cells had been generated from mesenchymal cells, bone marrow, adipose tissue, amniotic fluid, human umbilical cord blood, and skin 7 –16 . The skin, as well as the central nervous system (CNS), originates from the ectoderm, which might be an advantage to obtain neural precursors from epidermal cells. If epidermal cells from a skin biopsy of a patient can be induced to generate CNS derivatives, the induced neural cells will not elicit an immunological reaction if grafted in the same donor. In fact, fibroblasts have been genetically modified to generate iPSC 17 –19 that afterward can be differentiated into neurons (reviewed in Ref. 20 ). Furthermore, a direct conversion of phenotype from fibroblasts to neurons without an intermediate neural precursor state was reported after ectopic expression of transcription factors 21,22 .
The skin is an accessible source to obtain stem cells since the epidermis is an epithelium with continuous renewal. There are two populations of stem cells in the epidermis: one is located in the basal stratum and the second in the hair follicle’s protuberance (bulge). The potential for both populations to differentiate into neural lineages has been studied in rodents 23,24 and in humans 7,25,26 . It is possible to generate neurosphere-like aggregates from cultured dermal and epidermal rat cells, in the presence of a neural induction medium 23 . Likewise, the potential of human skin cells to differentiate into neural cells has been shown in vitro 25 . Furthermore, in vivo assays in adult organisms indicate that it is possible to differentiate rat cells derived from the skin into neural cells, when they are injected into both peripheral damaged tissue and in the spinal cord, leading to functional recovery 24 . However, it is not yet possible to generate neurons from epidermal cells efficiently.
Different protocols induce neuronal differentiation of NSCs; although they seem to function efficiently in mouse, human cells are not equally responsive. Experiments carried out in embryos, in which differentiation potential of cells was evaluated by grafting them in the CNS, have shown that the in vivo conditions provide an ideal environment to direct cell fate. Thus, this assay is useful to test the neural differentiation potential of non-neural cells. In this article, we established a procedure to isolate human keratinocytes from the epidermis, and cultured them in neural-favoring conditions to evaluate their differentiation potential when placed in the developing CNS of rat embryos. Our results show that it is possible to use ultrasound-guided transplantation of keratinocytes for embryonic xenografting of human cells, to determine if a neurogenic niche is capable to generate neural cells. We found that cells from the human epidermis can generate cells that express neuronal proteins in vivo.
Materials and Methods
Keratinocytes were obtained from the foreskin of males undergoing surgery, after donation through informed consent, at Hospital del Niño Morelense. Briefly, foreskins were treated with Dispase II (Roche Life Science, Penzberg, Germany) to detach the epidermis from the dermis. The epidermis was used to isolate keratinocytes and the remaining dermis was used to isolate fibroblasts. Before culturing them, some cells were set aside to perform immunophenotypic characterization. Keratinocytes were plated on tissue culture dishes with epidermal medium (UDMM-Epi, patent in progress). Cells were seeded at 1 × 106 in 55 cm2 at 37°C in a 100% humidified atmosphere with 5% CO2, reaching 80%–90% confluence after 4 days, when they were trypsinized and replated. Passage 3 cells were used for experiments.
Culture of Keratinocytes as Spheres
The initial density of the cultures was 2.65 × 105 cells/cm2. After 5 days, cells were detached from the tissue culture plate by trypsin treatment and cultured on ultra-low attachment six-well plates (Corning, New York, NY, USA). Two media were used at this stage: (a) control medium with 10% serum or (b) a serum-free medium with components regularly used for proliferating NSCs: Dulbecco’s modified Eagle’s medium (DMEM)-F12 with N2 supplement, 1% B27, 20 ng/ml epidermal growth factor (EGF), and 40 ng/ml basic fibroblast growth factor (bFGF). After 5 days, spheres were dissociated by trypsin treatment and reseeded on a tissue culture plate pretreated with poly-
Immunostaining
Attached cells were fixed with methanol for 20 min at −20°C. Spheres were let to settle and incubated with 4% paraformaldehyde in PBS and cut in the cryostat after cryoprotection with 30% sucrose. Cells were blocked with 10% normal goat serum plus 0.1% Triton X-100. Primary antibodies were added in a 10% goat serum solution. Spheres or dissociated cells were decorated with primary antibodies (source and working dilution indicated) recognizing KERATIN 5 (K5, Abcam, San Francisco, CA, USA, 1:100), KERATIN 10 (K10, Abcam, 1:100), KERATIN 14 (K14, Abcam, 1:100), NESTIN (Covance, Burlington, NC, USA, 1:100), SOX2 (Merck Millipore, Burlington, MA, USA, 1:500), VIMENTIN (Thermo, Waltham, MA, USA, 1:100), βIII-TUBULIN (TuJ1, Covance, 1:1,000), MICROTUBULE-ASSOCIATED PROTEIN 2 (MAP2, Sigma-Aldrich, 1:500), and glial fibrillary acidic protein (GFAP, Invitrogen, Carlsbad, CA, USA, 1:500). The secondary antibodies Alexa 488 goat anti-mouse and Alexa 568 goat anti-rabbit (Molecular Probes, Eugene, OR, USA) were used at 1:1,000 dilution. Positive controls for Sox2, Nestin, and βIII-Tubulin were performed with hESC differentiated to dopamine neurons as described 27 and are presented in Supplemental Figure 1.
Quantitative Real-time Polymerase Chain Reaction
Total RNA was isolated with TRIzol. cDNA was synthesized with 1 µg of RNA using random primers and SuperScript III Reverse Transcriptase (Invitrogen) at 37°C for 1 h. Amplification of 50 ng of cDNA was performed with the QuantiFast SYBR Green PCR Master Mix (Qiagen, Germantown, MD, USA) with a StepOnePlus Real-Time PCR System (Applied Biosystems, Waltham, MA, USA). Quantitative PCR was performed according to the following protocol: initial activation step for 5 min at 95°C. Two-step cycling: denaturation 10 s, 95°C. Combined annealing/extension: 30 s, 60°C (40 cycles). Ct values were analyzed using the StepOne software v2.3. The following specific primers (all in 5′ -> 3′ sequence) were used: NESTIN, forward (F): AGCCCTGACCACTCCAGTTTAG; reverse (R): CCCTCTATGGCTGTTTCTTTCTCT; product size of 128 bp. SOX2, F: AGCTACAGCATGATGCAGGA; R: GGTCATGGAGTTGTACTGCA; product size of 156 bp. VIMENTIN, F: CCAGGCAAAGCAGGAGTC; R: GGGTATCAACCAGAGGGAGT; product size of 138 bp. SOX1, F: GAGTGGAAGGTCATGTCCGAGG; R: CCTTCTTGAGCAGCGTCTTGGT; product size of 136 bp. MUSASHI1, F: GTCTCGAGTCATGCCCTACG; R: ACACGGAATTCGGGGAACTG; product size of 147 bp. β-ACTIN, F: GCTATCCAGGCTGTGCTATC; R: TGAGGTAGTCAGTCAGGTCC; product size of 166 bp.
Lentiviral Transduction with GFP and Selection by Fluorescent-activated Cell Sorting
Replication-deficient lentiviruses containing the cDNA for green fluorescent protein (GFP) under the EF1α promoter were prepared as described 28 , after transfection of HEK293 cells. Supernatants were collected after 48 and 72 h. The collected media were centrifuged at 27,000 rpm for 90 min at 4°C. The pellet was resuspended in 200 µl of sterile PBS, aliquoted, and kept at −70°C. Keratinocytes were transduced in serum-containing medium for 48 h. Three days after lentiviral addition, cells were dissociated and selected for high GFP expression by fluorescent-activated cell sorting (FACS) in a FACSAria equipment (BD, Franklin Lakes, NJ, USA). After selection by size and complexity, only cells that had values above 1 × 104 were recovered and plated on medium with serum. GFP-expressing cells were then cultured as spheres in either medium supplemented with serum or with the neural medium containing growth factors.
Ultrasound-guided Injection of GFP-positive Cells
All procedures in this study were conducted in accordance with the Instituto de Fisiología Celular-UNAM Animal Care and Use Committee (IVV67-15) approved protocols and followed National guidelines (NOM-062-ZOO-1999). Timed-pregnant Wistar rats with embryos of 12 days (E12) were used to perform ultrasound-guided injections of GFP-expressing cells similar to previous work 29 , with the aid of a MHF-1 Ultraview UltraSound system (E-Technologies, Davenport, IA, USA) possessing a focal distance of 7 mm. Briefly, deeply anesthetized dams (5% inhaled sevoflurane for induction and 1% throughout the procedure) were shaved and cleaned three times with 70% ethanol and iodopovidone. Midline sections of the skin and the muscle were performed to expose the uterine horns. Each embryo to be injected was secured by passing it through an incision made on rubber at the bottom of a petri dish. After the embryo passed, the dish was filled with sterile PBS and the ultrasound probe was placed close to the embryo. Injections were made through the uterine wall with borosilicate needles made with a puller (Sutter Instruments, Novato, CA, USA). The glass microcapillaries were visualized in the ultrasound imaging system and when in the lateral ventricles, injection of 3 µl of GFP+ cell suspension was corroborated by observing liquid going out of the needle after activating the automatic injector (Quintessential injector, Stoelting). Each embryo received only medium (control), or 4.5 × 105 cells from dissociated spheres cultured in either serum-containing or neural medium. Embryos were placed back in the abdominal cavity and the pregnant rat was sutured and let to recover. The embryos were recovered 4 days after the surgery, when they were at E16, placed with 4% paraformaldehyde in PBS overnight and transferred to 30% sucrose for cryoprotection. Sagittal sections of 30 µm were obtained in a cryostat; afterward, sections were blocked and incubated with anti-GFP and anti-βIII-Tubulin (TuJ1) or MAP2 antibodies. Appropriate fluorescent secondary antibodies were used to visualize the specific labeling. Sections incubated with secondary antibodies only did not present unspecific staining (data not shown). Confocal images were acquired with a Nikon A1 R HD25 microscope. All quantification results are given as mean ± standard error of the mean. Statistical analysis was made by paired Student’s t-test for comparison between two conditions or analysis of variance followed by Tukey post hoc for multiple conditions.
Results
To characterize foreskin human keratinocytes, we analyzed the expression of KERATIN 5, 10, and 14 (K5, K10, and K14, respectively) in isolated cells without culture. All these proteins were expressed in the analyzed cells (Fig. 1A–C). A low proportion of cells were positive for VIMENTIN (Fig. 1D) and NESTIN was absent (Fig. 1E). As expected, keratinocytes did not express DECORIN (Fig. 1F), but fibroblasts from foreskin were DECORIN-positive (Fig. 1G), supporting the notion that fibroblasts were absent in the isolated epidermis. Incubation of these cells with secondary antibodies did not show unspecific labeling (Fig. 1H). Quantification of the number of cells positive for these proteins showed that keratinocytes have a significantly higher proportion of K5, K10, and K14. In contrast, fibroblast have the selective expression of DECORIN and a higher proportion of cells positive for VIMENTIN (Fig. 1I).
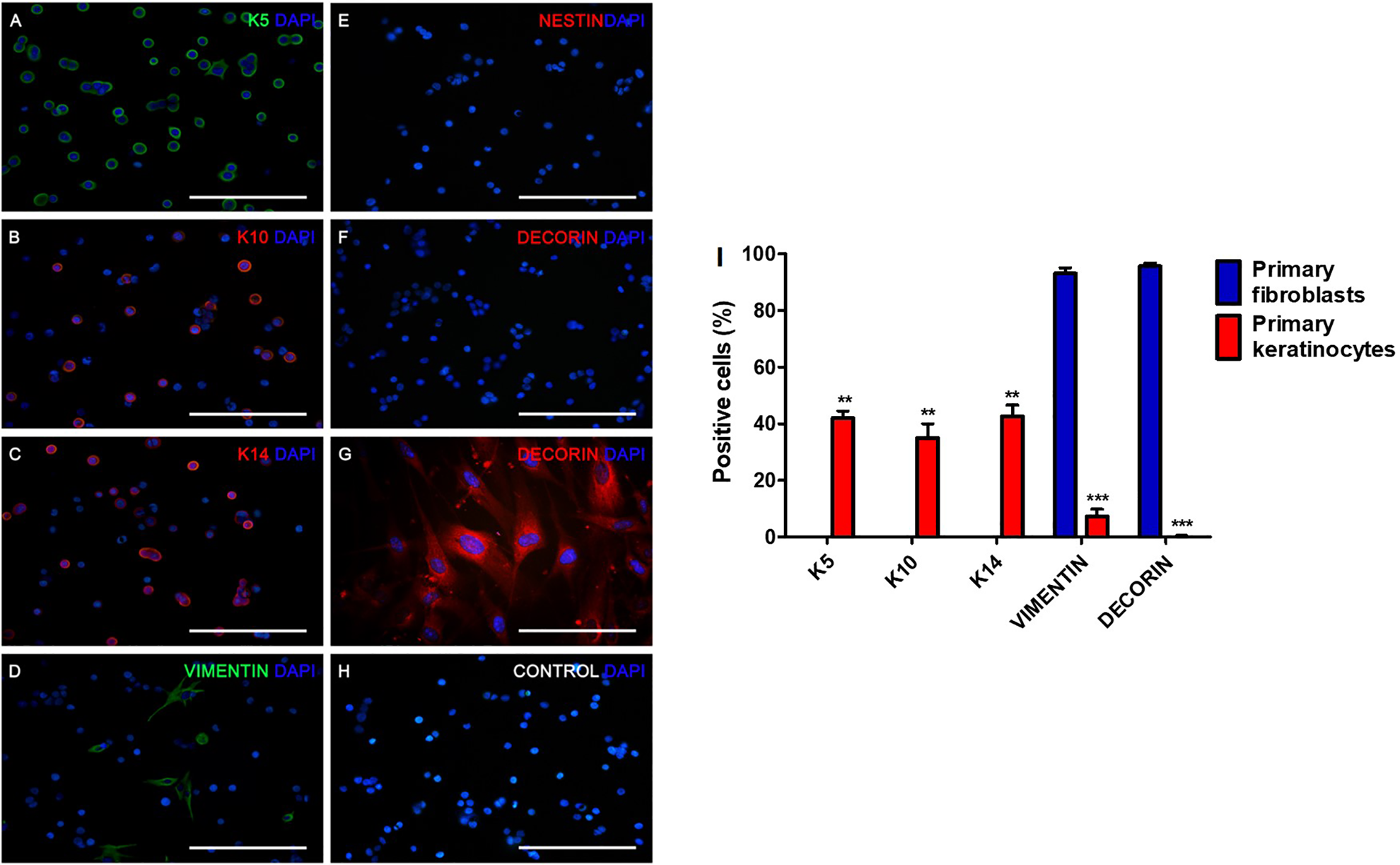
Isolated human foreskin keratinocytes before culture express KERATIN (K) 5 (A), K10 (B), K14 (C), and VIMENTIN (D). They were negative for NESTIN (E) and DECORIN (F). As a positive control for DECORIN, human fibroblast isolated from foreskin showed immunoreactivity (G). Cells incubated with only the secondary antibodies did not show unspecific staining (H). Scale bar = 15 μm. Keratinocytes selectively express K5, K10, and K14 compared with fibroblasts; fibroblasts presented exclusive positivity for DECORIN, compared with keratinocytes (I). **P < 0.01; ***P < 0.001.
Passage 3 keratinocytes were analyzed also for the expression of some of these markers: cells were negative for K10, but K5- and Vimentin-positive (data not shown). Keratinocytes at this passage were grown as aggregates in ultra-low attachment plates. Two different media were used, the serum-containing UDMM-Epi (control) and the serum-free neural medium. After 4 days, control cells produced larger spheres, when compared to the neural condition (Fig. 2A, B). After dissociation and replating, cells in the control medium showed the typical morphology of keratinocytes (Fig. 2C), whereas cells incubated with neural medium presented small and refringent somata with triangular shape and short processes, resembling a neural precursor-like morphology (Fig. 2D). At this stage, we investigated if expression of markers was modified by control or neural medium. By analyzing sections of spheres growing in control medium, we only observed VIMENTIN-positive cells; aggregates were negative for NESTIN (Fig. 3A). In contrast, the neural medium with EGF and bFGF did not affect the expression of VIMENTIN, but significantly induced the expression of NESTIN (Fig. 3B, C). These spheres were dissociated, replated, and stained just after attachment. Cells in the control medium expressed K5 and Vimentin, but not K10, K14, or NESTIN (Fig. 4A). Cells incubated in the neural medium with growth factors were negative for K5, K10, and K14, preserved Vimentin and started to express NESTIN (Fig. 4B). The neural conditions caused significant decrease of K5+ cells and also a significant increase of NESTIN-positive cells (Fig. 4C). Quantitative PCR with primers specific for NESTIN, SOX2, VIMENTIN, SOX1, and MUSASHI1, after retrotranscription, showed that the neural conditions significantly increased expression of these transcripts, relative to cells growing in the control medium with serum, either in spheres or attached in monolayer (Fig. 5).
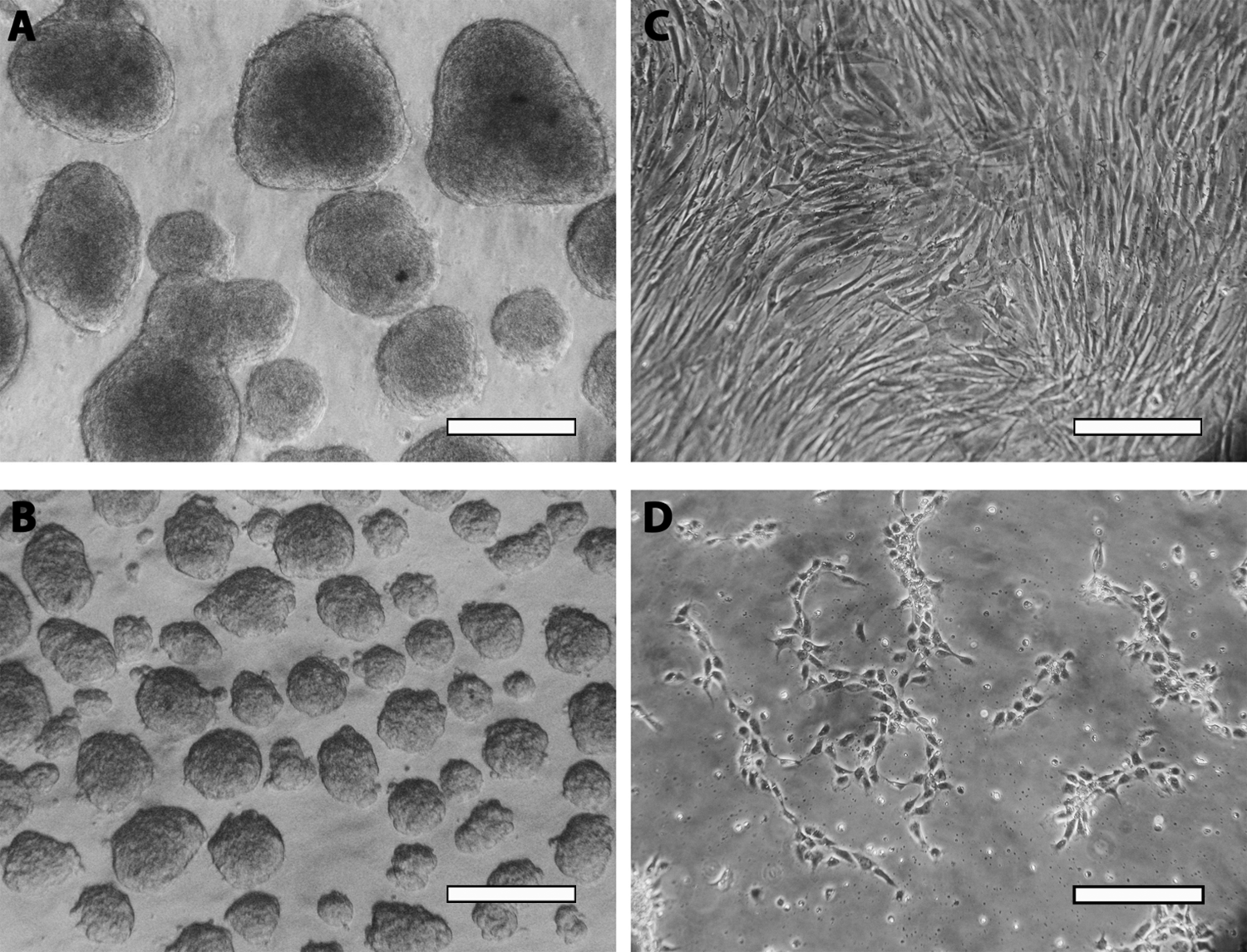
Keratinocytes cultured as aggregates in UDMM-Epi serum-containing medium (A) or neural medium (B). After 4 days, aggregates were dissociated and replated on tissue culture plates either in UDMM-Epi (C) or in neural medium (D). Note that the aggregates in UDMM-Epi are larger and dissociated cells in UDMM-Epi show classical keratinocyte morphology. In contrast, dissociated cells incubated in neural medium present morphology reminiscent of neural precursors. Scale bar = 150 µm (A and B) and 50 µm (C and D).
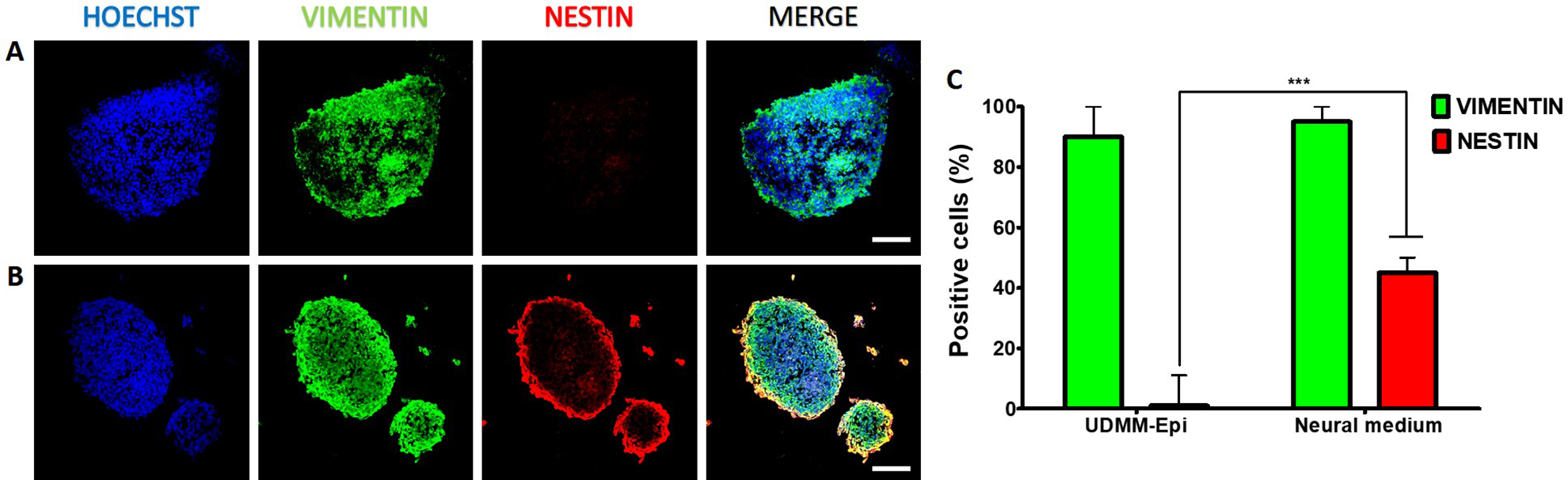
Aggregates either in UDMM-Epi medium (A) or in neural medium (B) were fixed, sectioned, and stained for VIMENTIN and NESTIN. Scale bar = 50 μm. The neural medium significantly induced the presence of NESTIN-positive cells (C). ***P < 0.001.
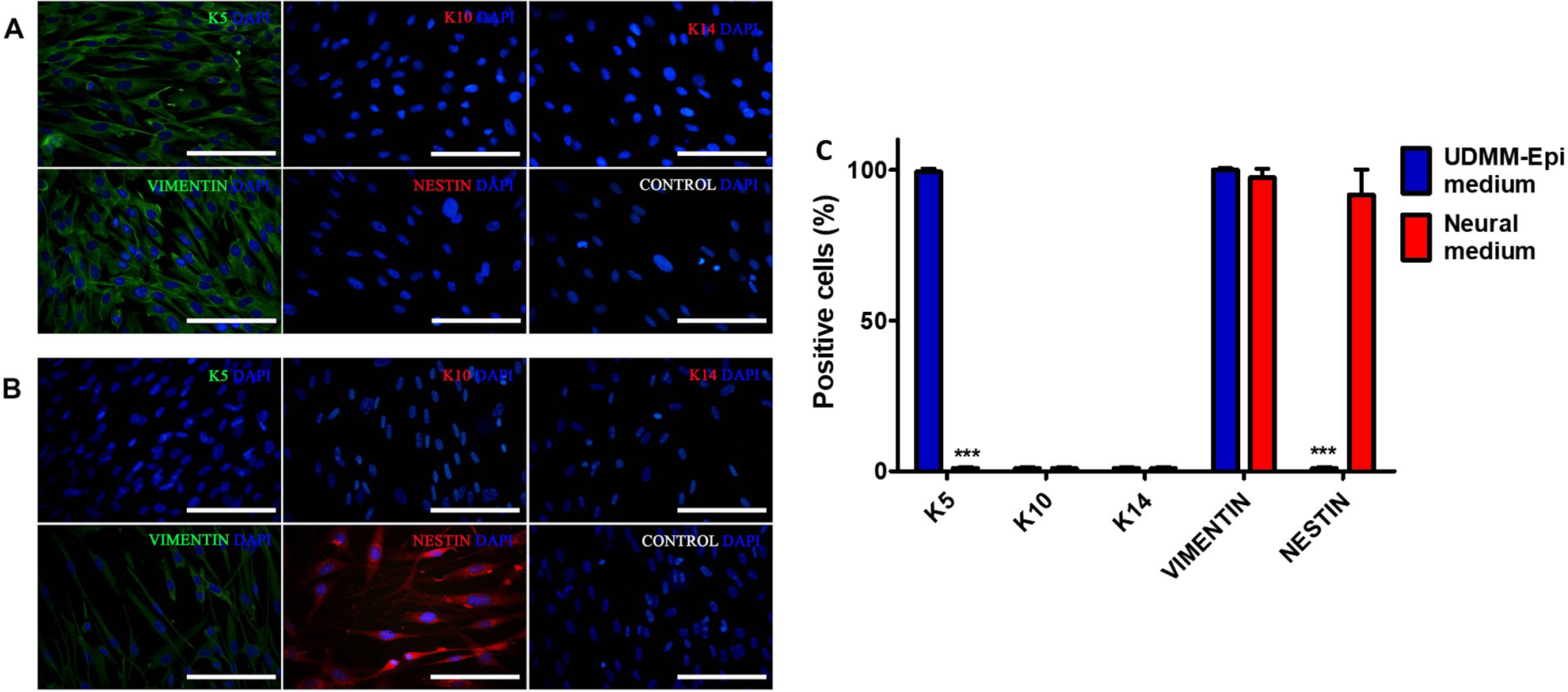
Aggregates were dissociated, replated, and fixed to analyze the expression of several markers. Keratinocytes growing in UDMM-Epi medium (A) were positive for K5 and VIMENTIN, and negative for K10, K14, and NESTIN. Cells cultured in the neural medium (B) lost the expression of K5 and gained NESTIN immunoreactivity. In both panels, control pictures correspond to cells incubated only with secondary antibodies, to discard unspecific binding. Scale bar = 15 μm. Cells in control medium had a significantly higher proportion of K5+ cells and the neural medium significantly increased the presence of NESTIN-positive cells (C). ***P < 0.001.
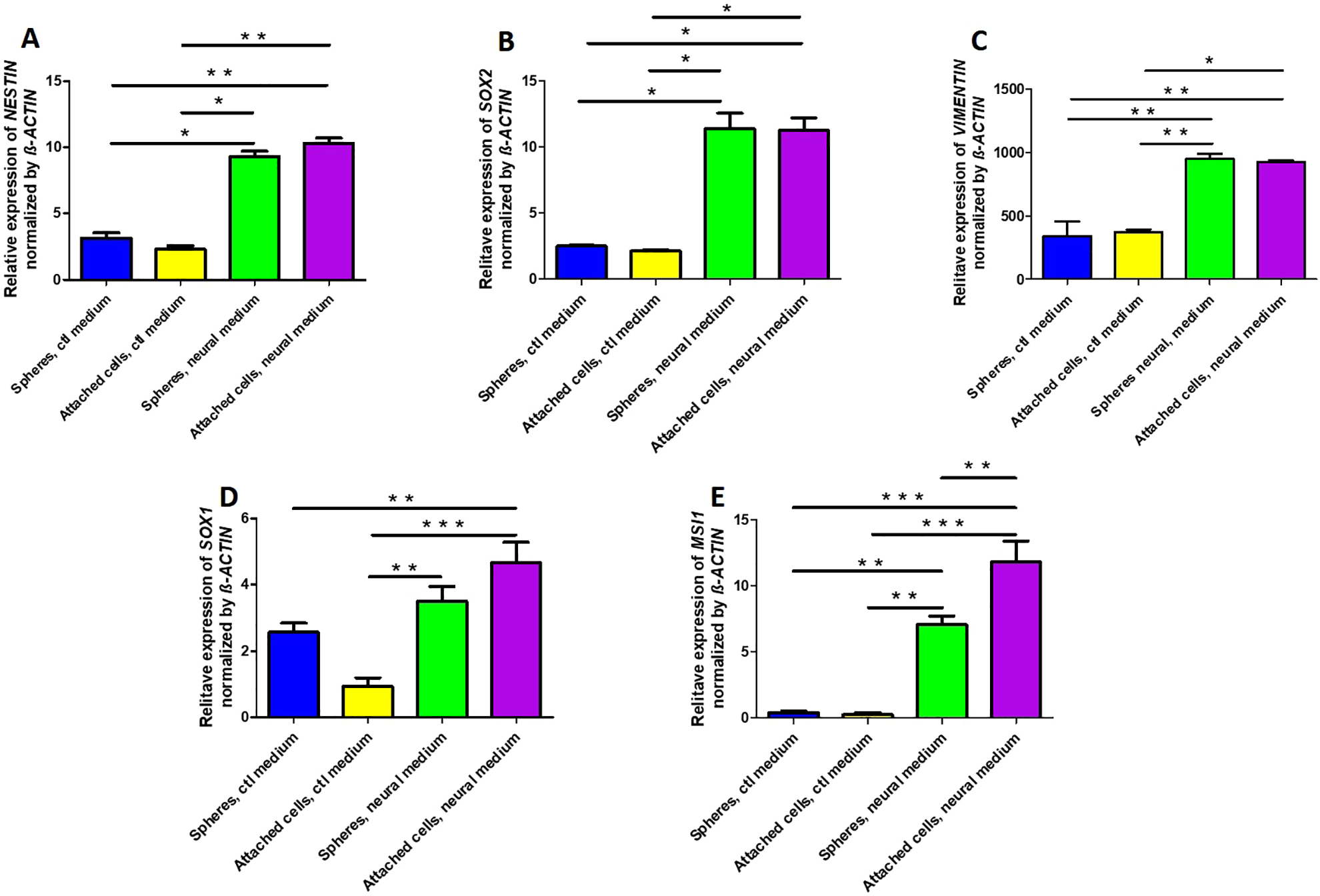
Analysis of expression of transcripts for NESTIN (A), SOX2 (B), VIMENTIN (C), SOX1 (D), and MUSASHI1 (MSI1, E) by RT-qPCR in the serum containing (Control, ctl) or the neural medium in cells growing as aggregates or after dissociation and replating. Total RNA was isolated from the indicated conditions and retrotranscribed to obtain cDNA, which was amplified by qPCR with specific primers. These values were normalized by the housekeeping gene β-ACTIN. Bars are mean ± standard deviation. *P < 0.05; **P < 0.01; ***P < 0.001. RT-qPCR: quantitative real-time polymerase chain reaction.
Attached cells were cultured for 4 days after dissociation of spheres, and immunostainings for the astrocytic marker GFAP and the neuronal protein βIII-Tubulin (TuJ1 antibody) were performed, but cells did not express these differentiation markers. To test if the in vivo environment of the developing brain can instruct cells cultured in epidermal or neural conditions to differentiate into neural phenotypes, intrauterine ultrasound-guided transplantation was performed in brain of rats. To unequivocally identify grafted cells, keratinocytes were transduced with lentiviral vectors to achieve constitutive expression of GFP. Although a good proportion of cells were fluorescent after transduction (Fig. 6A), we decided to select the high GFP-expressing cells by FACS. For this purpose, cells were dissociated and sorted in a FACSAria I cytometer. Individual cells were selected (Fig. 6B), cellular debris was excluded (Fig. 6C), and 23% of cells with values over 1 × 104 were recovered. After plating, all cells were GFP+ (Fig. 6E). These GFP keratinocytes were cultured in either serum-containing medium (Fig. 6F) or neural medium (Fig. 6G) and showed a similar behavior to untransduced keratinocytes in sphere formation; all cells remained positive for GFP.
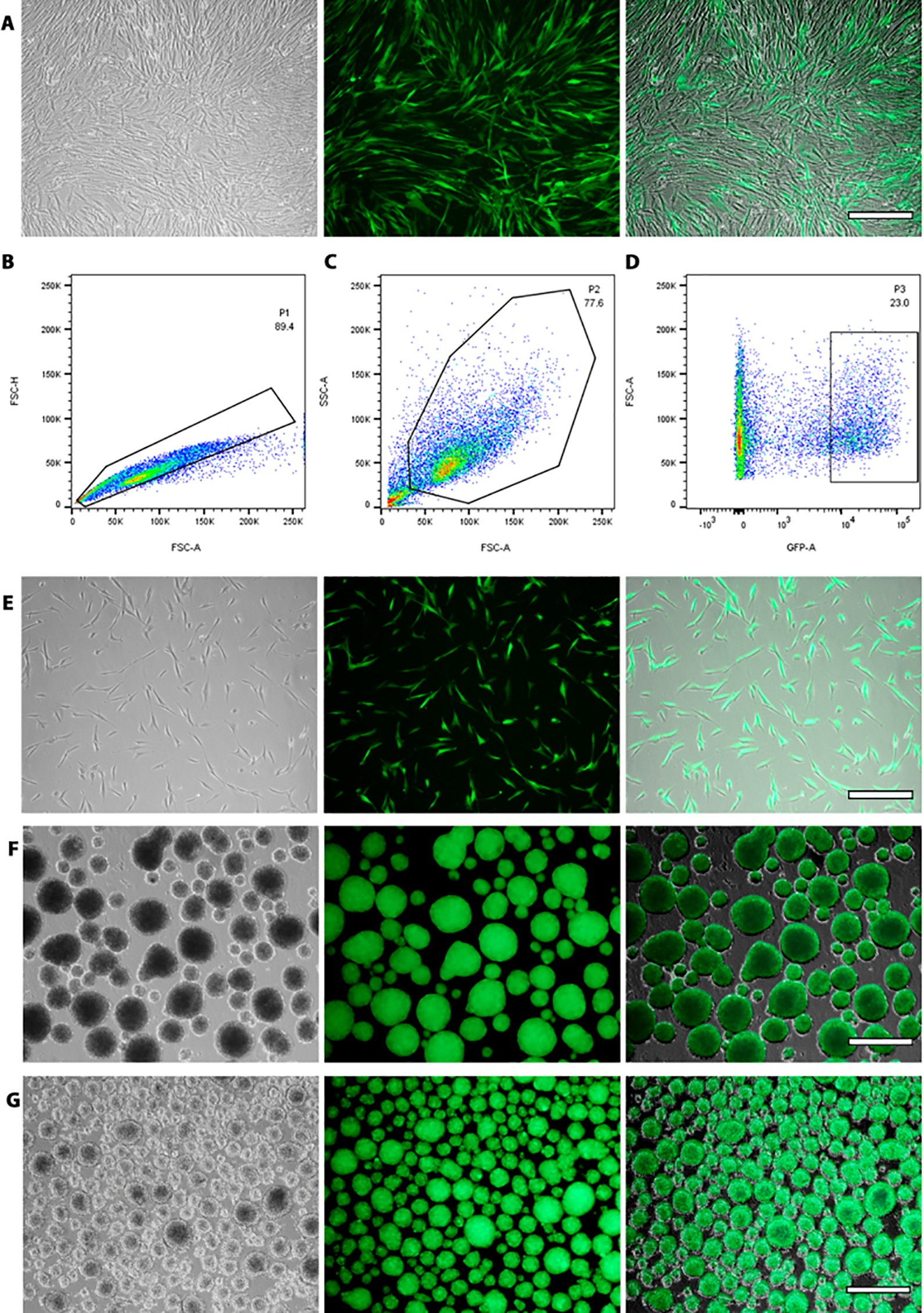
Isolation of GFP-expressing keratinocytes after lentiviral transduction. (A) Keratinocytes growing in UDMM-Epi medium after viral transduction. Cells were trypsinized and analyzed by flow cytometry. Individual cells were selected (B), cellular debris was omitted (C), and cells with a high value of GFP fluorescence were recovered by cell sorting (D). All cells were GFP-positive after sorting (E). These fluorescent cells were grown as aggregates in UDMM-Epi (F) or neural (G) media. For A, E, F, and G, the images on the left correspond to phase contrast, the middle panel to the fluorescent picture, and the right picture is the merge of the former 2. Scale bar = 50 µm (A and E) and 150 µm (F and G). GFP: green fluorescent protein.
Ultrasound-guided intrauterine injection in the lateral ventricles of E12 embryos was performed next. As a control, only DMEM was introduced. The analysis showed no alterations in the brain of these embryos 4 days later (data not shown). The brains of embryos injected with GFP+ cells were recovered and sectioned. After injection of dissociated spheres grown in control medium, we found that three out of three embryos had GFP-positive cells that expressed the neuronal markers βIII-Tubulin, detected with the TuJ1 antibody, and MAP2 (Fig. 7A), although their morphology was immature, which can be appreciated in the high-magnification confocal images, that also show colocalization of GFP and the neuronal proteins in the orthogonal projections. Cells cultured as spheres in the neural medium were dissociated and grafted in the developing brain; we found GFP+ cells that coexpressed βIII-Tubulin and MAP2, with a more intricate morphology, pointing to differentiation into neurons (Fig. 7B) in two out of three injected embryos. The number of GFP-positive per brain slice was similar in both groups: control medium, 79.7± 11.6; neural medium, 57 ± 11.4.
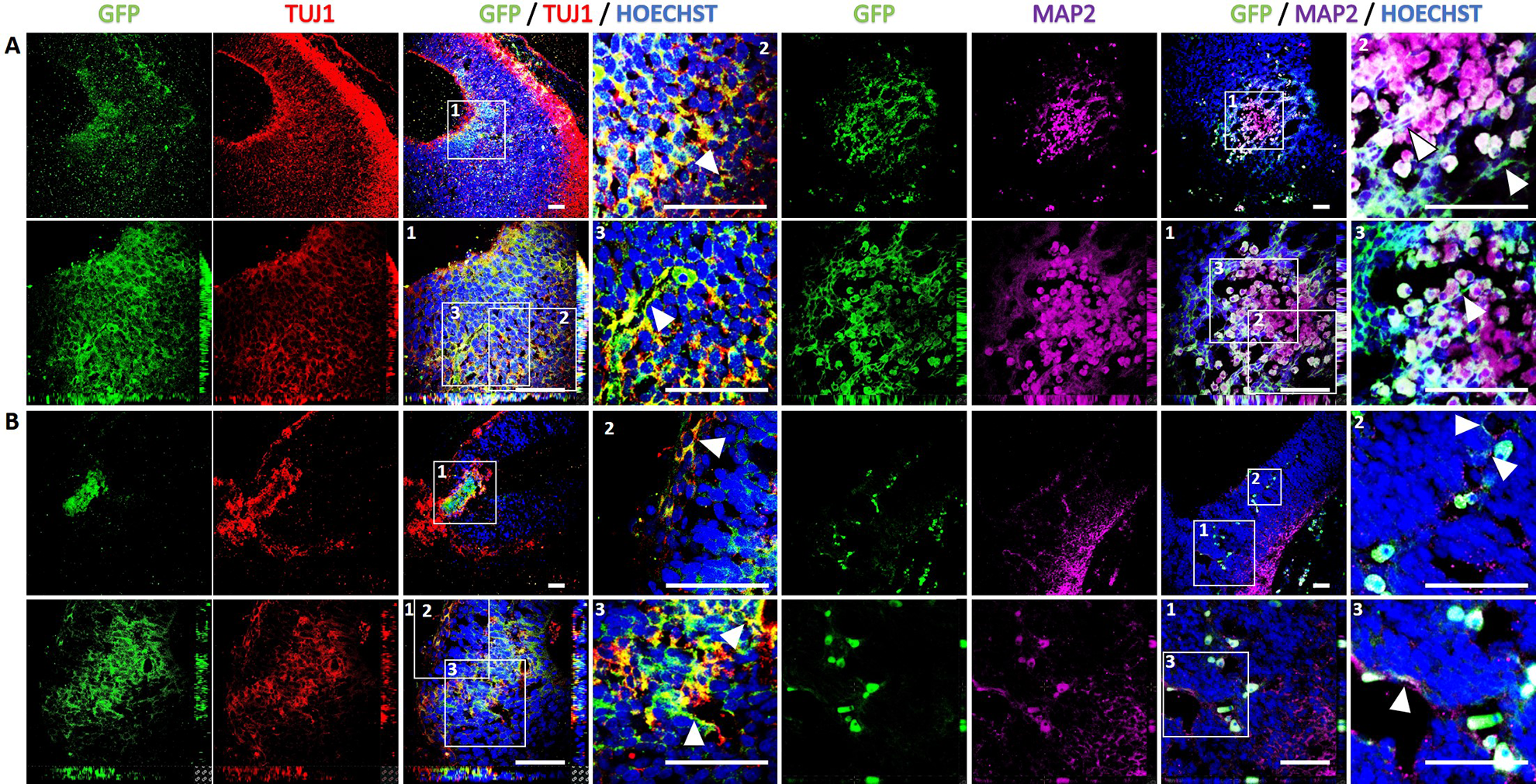
Human keratinocytes differentiate to neuronal-like cells when grafted in the developing rat brain. Ultrasound-guided injections of dissociated aggregates cultured in UDMM-Epi medium (A) or neural medium (B). Grafting was performed in 12-day-old embryos and the analysis was performed 4 days later in the brain of the recovered embryos. GFP is shown in green, β-III TUBULIN (detected with the TUJ1 antibody) is presented in red, Hoechst staining of the nuclei is shown in blue in the merge images, and MAP2 in magenta. The top row of each panel corresponds to low-magnification confocal images, and the third picture corresponding to the merge image. A white square labeled 1 marks the area shown at higher magnification in the second row images of each panel, that include orthogonal projections. To better appreciate the neuronal-like morphology, zoom-in images labeled with white squares numbered 2 and 3 are presented in the fourth column for each neuronal marker. The GFP-positive processes of cells that coexpress β-III TUBULIN or MAP2 are indicated with white arrowheads. Scale bars = 50 μm.
Discussion
Conversion of cell phenotypes from different sources has been reported by ectopic expression of transcription factors or molecules added to specific culture media. We report that we can convert/transdifferentiate human keratinocytes into neural stem/progenitor-like cells (NSPCs) in vitro just by culturing them with a neural media in the presence of EGF and bFGF, even though this was not enough to generate differentiated neurons. Our research work was focused on the generation of neural cells from human epidermal cells. Keratinocytes were cultured in favorable conditions for their propagation with the UDMM-EPi medium, for testing the generation of NSPC. A neural medium was employed, combined with culture in ultra-low-adherence conditions, in the presence of the mitogens EGF and bFGF. Under such conditions, cells started to upregulate transcripts associated to NSPCs, such as SOX2, NESTIN, SOX1, and MUSASHI1. Although it was possible to generate aggregates from keratinocytes on UDMM-Epi medium, they did not express NESTIN, a protein present in aggregates cultured in neural medium. However, when the aggregates were dissociated and transplanted to the lateral ventricles of E12 rat embryos, cells cultured in UDMM-Epi or neural medium demonstrated their capacity to generate neurons. Thus, human keratinocytes are able to generate in vivo cells positive to β-III TUBULIN and MAP2, showing that the microenvironment present in the developing CNS was optimal for evaluating their complete differentiation potential.
Molecular Markers Expressed by Keratinocytes in Culture
Keratinocytes make up the epidermis: the proliferating precursor cell population is located in the basal layer, and differentiated cells form the suprabasal (spinous, granular, and corneum) layers 30,31 . The different populations are distinguished from each other by their Keratin expression patterns 32 . When keratinocytes exit cell cycle to start differentiation, they decrease the expression of K5 and K14 33 , and increase the expression of K1 and K10 31 . Keratinocytes isolated here show the expected pattern of basal and early differentiating layers (K5, K10, and K14) at low passages, although we also found a subpopulation that was not fibroblasts, expressing VIMENTIN (Fig. 1). The proportion of cells that express VIMENTIN increased by passage 3, concomitant with loss of K14 and K10; however, they remained positive K5 and VIMENTIN (Fig. 4), and maintained a high proliferative rate, since they reached confluence in 4 days. The in vitro conditions established by our group maintain proliferative cells for up to 20 passages (data not shown) with sustained expression of K5.
Generation of Cells with Some Neural Stem/Progenitor Characteristics from Keratinocytes
In vitro propagation of NSPCs was described by pioneering reports of adult neural tissue in rodents 3,34 and subsequently in human tissue 35 . These cultures included the mitogenic factors EGF and bFGF and were designated neurospheres, because they grew in suspension 36 and included the mitogenic factors EGF and bFGF. Such conditions were used afterward in the propagation of NSPCs from embryos 34,37 . Based on this method, the generation of neural precursors from adipose 13,14 , mesenchymal 7,12 , and epidermal 11,15,16 cells were reported. While it has been possible to generate neurons, the efficiencies using human cells have been low: only 5% of the cells showed neuronal differentiation using spheres from adult dermis and up to 15% when embryonic tissue was used 7 . Different results have been described for the tissue obtained from rodents, where it was possible to obtain up to 48% of neurons from bulge stem cells 38 . Recently, it was reported that neural crest stem cells can be generated from human interfollicular and epidermal keratinocyte cultures, supporting the relevance of bFGF in neural specification 39,40 . In the present work, using conditions for the production of neurospheres, we generated human NSPCs, identified by the expression of the well-recognized NSPCs markers SOX2, NESTIN, SOX1, and MUSASHI1 41,42 , from keratinocytes.
In vitro assays to demonstrate neuronal differentiation of our aggregates were unsuccessful. Nevertheless, it has been demonstrated that transfer of cells obtained from human neurospheres can integrate and differentiate to adult neural tissue of rodents and generate immature neurons 7 . Noteworthy, the processes of differentiation and determination in the adult CNS are different from those present during embryonic organogenesis 43 . Recently, embryonic brain extracts showed to favor neural differentiation of mesenchymal stem cells obtained from rat bone marrow 44 .
Spheres Derived from Skin and Their Neural Differentiation Potential
The conditions used for the propagation of NSCs include incubation with bFGF and EGF 3 , which have been used to induce the formation of neurospheres from different tissues and tumors 45 . Such neurospheres are formed after cell proliferation, producing cell aggregates that can be of clonal origin. In our conditions of low attachment and high density, aggregation and proliferation contribute to sphere formation and growth. It has been reported that clonal generation of epidermal spheres takes 2 to 3 weeks 46 . Different groups have isolated spheroid structures from the skin, which include the dermis 10,47 –49 , dermal papilla 50 , hair follicle outer root sheath cells 51 , hair follicle 11 , and epidermis 46 with the potential for neural differentiation. The experimental conditions that have been reported are very similar to those used by our group, particularly defined medium in the presence of bFGF and EGF. Notably, most of the spheres generated from skin have been related to neural crest lineages, given their differentiation toward Schwann cells and/or smooth muscle cells 10,11,48,49 . A report used epidermal cells, but further characterization established that it was a population of the outer sheath of the follicle, since cells were positive for Fibronectin, Nestin, and CD34 46 and thus also related to neural crest 47,49 . One of these papers reported two types of stem populations isolated from the skin, one adherent, of mesenchymal origin, and the second in suspension with neural crest origin 49 . The characterization that we carried out in our initial population (positivity for K5, K10, and K14) established that it was obtained from the interfollicular epidermis, and did not contain fibroblasts. The neural differentiation of the epidermal spheres has been evaluated in transplantation experiments in postnatal rats, using cells that incorporated the Hippel-Lindau tumor suppressor protein. They found that 38.8% of cells differentiated to TUJ-1 positive cells in vivo 46 . In comparison, our experimental model uses fetal transplantation of cells in the developing brain, a neurogenic environment, in the absence of genetic manipulations, which might represent a more appropriate system to study plasticity events.
The Effect of the Developing CNS Environment on Neuronal Differentiation
Skin/epidermal cells and neural cells share the same embryonic origin, the ectoderm. The fetal brain possesses a robust plasticity and holds a strong neurogenic environment. Previously, it has been reported that specific embryonic brain regions such as midbrain, in explants, are able to direct neuronal specification of neural precursor cells as well as undifferentiated embryonic stem cells and embryoid bodies 52,53 .
In early work, adult rat mesenchymal cells were labeled with BrdU and injected in the ventricles of E15.5 rat brains. Grafted cells remained as groups close to the ventricles, presumably in the germinal zones, and expressed Vimentin and Nestin, but not neuronal markers. Above 50% of the grafted cells were positive for the radial glia marker RC2. Terminally differentiated neurons were found after birth in the cerebral cortex and the midbrain up to 2 months of age 54 . In contrast, amnion-derived stem cells labeled with GFP were injected at E15 rat cerebral ventricles. Grafted cells were present in adult organisms, but no evidence of neuronal or astrocytic differentiation was reported; interestingly, these cells can generate endothelial cells in the host brain 55 . With regards to human cells, MSC obtained from adult brains (white matter and hippocampus) have been tested after transplantation in E10 mice. After 7 days, only 20% of the brains presented donor cells, which clustered in the ventricular system. Grafted cells, identified by the Human Nuclear Antigen, were negative for Sox2 (neural precursors), Doublecortin (immature neurons), and MAP2 (mature neurons); only a few were positive for GFAP, indicating astroglial differentiation 56 . Recently, a method using hESCs differentiated to cortical progenitors allowed robust engraftment in mouse E14.5 brains. One day later, Pax6-positive human cells were found in the ventricular zone, but at later time points, they reached the cortical plate. These βIII-Tubulin+ neurons presented a polarized morphology and they mimicked the radial migration characteristic of the cerebral cortex. Intriguingly, these neurons were found throughout all cortical layers at postnatal day 7, but nonexistent in postnatal day 28 57 . We have found conditions to induce in vitro the expression of the NSPC markers SOX2, NESTIN, SOX1, and MUSASHI1 by human keratinocytes. Interestingly, both induced cells and keratinocytes that did not express such markers were able to produce neurons after transplantation in the developing rat brain.
Conclusions
Here, we report that human keratinocytes are able to transdifferentiate/convert into differentiated neuronal-like cells positive for β-III TUBULIN and MAP2 when grafted in the developing brain in vivo, supporting the presence of a permissive or an inductive neurogenic environment even for cells not committed to differentiate into neural phenotypes such as keratinocytes. These findings suggest that the skin might be used as a relevant source of neurons in conditions where cell replacement is required. It remains to be analyzed the integration of converted keratinocytes into specific neuronal circuits and the neuronal phenotypes that might be generated.
Supplemental Material
Supplemental Material, sj-tif-1-cll-10.1177_0963689720978219 - Human Keratinocytes Adopt Neuronal Fates After In Utero Transplantation in the Developing Rat Brain
Supplemental Material, sj-tif-1-cll-10.1177_0963689720978219 for Human Keratinocytes Adopt Neuronal Fates After In Utero Transplantation in the Developing Rat Brain by Andrea Tenorio-Mina, Daniel Cortés, Joel Esquivel-Estudillo, Adolfo López-Ornelas, Alejandro Cabrera-Wrooman, Rolando Lara-Rodarte, Itzel Escobedo-Avila, Fernanda Vargas-Romero, Diana Toledo-Hernández, Enrique Estudillo, Juan José Acevedo-Fernández, Jesús Santa-Olalla Tapia and Iván Velasco in Cell Transplantation
Footnotes
Acknowledgments
We thank Dr Jessica Marin and Dr Jesús Chimal-Monroy for assistance on cell sorting. We acknowledge Eugenio López Bustos and Jorge Arturo Yañez Ponce de León for their technical assistance.
Ethical Approval
Ethical approval for the use of the foreskins was obtained from the Research Committees at Hospital General de Cuernavaca “José G. Parres”, Hospital del Niño Morelense, and Facultad de Medicina Universidad Autónoma del Estado de Morelos, Cuernavaca, Morelos, México (authorization numbers JE/730/04, E.I./05/278, study No. 2017-76).
Statement of Human and Animal Rights
All animal procedures used in this study were approved by the Instituto de Fisiología Celular-UNAM Animal Care and Use Committee (Protocol IVV67-15) and followed National guidelines (NOM-062-ZOO-1999). This article does not contain any studies with human subjects.
Statement of Informed Consent
There are no human subjects in this article, but written informed consent was obtained from a legally authorized representative for foreskin use. The form was approved by the Research Committees from Hospital General de Cuernavaca “José G. Parres”, Hospital del Niño Morelense, and Facultad de Medicina Universidad Autónoma del Estado de Morelos, Cuernavaca, Morelos, México.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was partially supported by Papiit-UNAM (IN213716 and IN213719), CONACYT (139851, 151287, 241374, 300461, and Red Temática de Células Troncales y Medicina Regenerativa), Rubio Pharma y Asociados S.A. de C.V., and PROMEP (CA-75).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.