
Abstract
Since the 1970s, rodent and human insulin-secreting pancreatic beta-cell lines have been developed and found useful for studying beta-cell biology. Surprisingly, although the dog has been widely used as a translational model for diabetes, no canine insulin-secreting beta cells have ever been produced. Here, a targeted oncogenesis protocol previously described by some of us for generating human beta cells was adapted to produce canine beta cells. Canine fetal pancreata were obtained by cesarean section between 42 and 55 days of gestation, and fragments of fetal glands were transduced with a lentiviral vector expressing SV40LT under the control of the insulin promoter. Two Lox P sites flanking the sequence allowed subsequent transgene excision by Cre recombinase expression. When grafted into SCID mice, these transduced pancreata formed insulinomas. ACT-164 is the cell line described in this report. Insulin mRNA expression and protein content were lower than reported with adult cells, but the ACT-164 cells were functional, and their insulin production in vitro increased under glucose stimulation. Transgene excision upon Cre expression arrested proliferation and enhanced insulin expression and production. When grafted in SCID mice, intact and excised cells reversed chemically induced diabetes. We have thus produced an excisable canine beta-cell line. These cells may play an important role in the study of several aspects of the cell transplantation procedure including the encapsulation process, which is difficult to investigate in rodents. Although much more work is needed to improve the excision procedure and achieve 100% removal of large T antigen expression, we have shown that functional cells can be obtained and might in the future be used for replacement therapy in diabetic dogs.
Introduction
The incidence of insulin-dependent diabetes (IDDM) in dogs is similar to that in humans and has increased recently 1,2 . Just as in humans, IDDM in dogs is due to selective beta-cell destruction within the pancreatic islets 3 , causing severe insulin deficiency. Given the numerous similarities of the disease between humans and dogs, several authors 4 –6 have underlined the importance of studying dogs with IDDM, not only to improve the condition in the animals but also as a model for advancing diabetes care in humans.
Our objective here was to develop an immortalized, functional, canine beta-cell line. Having an abundant source of canine beta cells would not only allow transplantation as replacement therapy for canine diabetes but also provide a model for investigating many aspects of beta-cell transplantation. For example, such a model would allow the testing of several aspects of the encapsulation process, which is critical to the success of beta-cell transplantation 7 . We believe that a canine beta-cell line will offer new treatment prospects for canine diabetes while also helping to build a model that more closely resembles human diabetes and that will serve to achieve advances in the transplantation procedure in humans.
Much effort has been expended in the last 20 years to produce engineered human beta-cell lines for cell therapy in diabetes. Human embryonic stem cells and induced pluripotent stem cells can both be directed to differentiate into pancreatic progenitors that mature into functional, glucose-responsive, insulin-secreting cells. Although this procedure holds considerable promise, major improvements are still needed to increase cell production yields and cell maturity 8,9 . We have previously reported the production of rat and human beta-cell lines generated from fetal pancreases by targeted oncogenesis and then differentiated in vivo in mice 10,11 . These cells are immortalized and continue to proliferate, unlike primary beta cells. To prevent this continuous proliferation, we have devised a strategy to generate conditionally immortalized human beta-cell lines based on Cre-mediated excision of the immortalizing transgenes 12 . Transgene excision resulted not only in an arrest of cell proliferation but also in pronounced enhancement of beta cell-specific features such as insulin expression and content. This technique has been developed further and adapted to the dog to produce canine beta cells. We report here the successful production of a canine beta-cell line named ACT-164. These cells are functional, and removing the immortalizing transgene increases the production of insulin. To our knowledge, this is the first canine beta-cell line available for the development of preclinical models.
Materials and Methods
Animals
Pancreas collection was conducted at the Maisons-Alfort Veterinary School (facility 947-046-2), with the approval of the national review board CNREEA #16 (APAFIS #2015042112442132). The procedures for tissue and cell transplantation in mice were approved by the French Ministry of Research and Education (#01592.02). Our methods complied with international regulations for the use of animals in experimental studies. Male SCID mice aged 6 to 8 weeks (CB-17/Icr-Prkdc scid/Rj, Janvier Labs, Le Genest-Saint-Isle, France) were used in all protocols.
DNA Constructs and Lentiviral Vector Production
The lentiviral construct pTRIP ΔU3.RIP405-SV40 LT has been described elsewhere 10 –13 . The new lentiviral vector pTRIP ΔU3 loxP.RIP405-SV40 LT was constructed. The loxP 3′LTR cassette was amplified by polymerase chain reaction (PCR) from the SIN-loxP vector (kindly provided by B. Thorens, Lausanne University, Lausanne, Switzerland) with the Kpn primer 5′-CGGGGTACCTTTAAGACCAATGACTTACA-3′ and the PacI primer 5′-CGGTTAATTAAGAACTACTACTGCTAGA-3′. The resulting 378-pb PCR fragment was digested by KpnI and PacI restriction endonucleases and then inserted into the SV40 LT lentiviral vector to replace the ΔU3 3′LTR by a loxP 3′LTR. The pTRIP ΔU3 CMV-nlsCRE has been described elsewhere 14 .
Lentiviral vector stocks were produced by transient transfection of HEK 293T cells by encapsidation of the p8.9 plasmid (ΔVprΔVifΔVpuΔNef) pHCMV-G encoding the VSV glycoprotein-G and the pTRIP ΔU3 recombinant vector, as previously described 15 . The supernatants were treated with DNAse I (Roche Diagnostics, Basel, Switzerland) prior to their ultracentrifugation, and the resultant pellets were resuspended in phosphate-buffered saline (PBS), aliquoted, then frozen at −80 °C until use. The amount of p24 capsid protein was quantified by the HIV-1 p24 antigen ELISA (Beckman Coulter, Brea, CA, USA). All transductions were normalized relative to the amount of p24 capsid protein.
Canine Pancreatic Tissue Collection
All antenatal samples were obtained from a strain of Beagle dogs raised at the Maisons-Alfort Veterinary School. Specimens were obtained by elective cesarean section of pregnant dogs. Fetal age was determined based on the time of the plasma progesterone surge indicating ovulation. Different specimens were collected at different times between 41 and 55 days of gestation according to the plasma progesterone peak. Each pancreas was dissected within 1 h after the cesarean section and prepared for grafting. Depending on the size of the litter, 4 to 6 pancreata were collected.
Tissue Treatment Before Transplantation
Fetal pancreata were minced in small fragments less than 1 mm3 and treated for 15 min at room temperature with 0.03% collagenase A (Roche, Basel, Switzerland) solution in Roswell Park Memorial Institute (RPMI) 1640 (Life Technologies, Villebon sur Yvette France). Collagenase digestion was stopped in ice-cold Hanks’ balanced salt solution (HBSS). Such treatment allows partial digestion of pancreatic tissue and increased tissue accessibility to the lentiviral vector. Such pretreated tissues were either transplanted immediately or transduced with the lentiviral vector before transplantation.
Gene Transfer
Partially digested pancreata were transduced with pTRIP ΔU3 loxP.RIP405-SV40 LT using a total amount of lentiviral vectors corresponding to 2 µg of p24 capsid protein, for 2 h at 37 °C, in 200 µl of RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; 10 mM),
Fetal Pancreatic Tissue Transplantation into SCID Mice
Pancreatic specimens collected between 41 and 55 days of gestation were partially digested, transduced or not, and transplanted under the kidney capsule as described previously 11 . Briefly, the left kidney was exteriorized; a small transverse incision was made through the capsule on the ventral surface of the kidney, near the inferior pole. A silicon O-ring (2.5 mm inner diameter) was pushed under the capsule to provide a sealed space to confine the transplanted tissues. Pancreatic fragments were simply placed in the middle of the O-ring using forceps.
In the first step, we assessed the survival and cell composition of both untransduced and transduced canine fetal pancreata. Mice were sacrificed 2 (n = 2), 4 (n = 2), and 6 (n = 5) months after transplantation. After dissection, the transplant was prepared for morphological examination and localization of alpha and beta cells by immunohistochemistry.
In the second step, grafts issued from several transduced pancreata were then removed after 6 to 9 months in order to derive cultured immortalized cell lines.
Immortalized Cell Line Derivation
SCID mice grafted with transduced pancreatic tissues were sacrificed 6 to 9 months after transplantation. The grafts were removed, cut into pieces in a sterile cabinet, and weighed. The graft fragments were treated with 200 units of type IV collagenase in 500 ml of HBSS for 15 min at 37 °C. The digested tissue was centrifuged for 8 min at 3000 rpm, and the resulting pellet was resuspended in a solution containing PBS 1× and 25% decomplemented fetal bovine serum. The suspension was mechanically dissociated in a 1 ml syringe by successive passages through 20, 23, 25, 27, and 30-gauge needles. The dissociated cells were centrifuged for 5 min at 3000 rpm and resuspended in a volume of 500 µl in the solution described above. After this step, the cells were centrifuged again for 5 min at 3000 rpm and then resuspended, this time in RPMI 1640 culture medium containing 10% of decomplemented fetal bovine serum and 1% of penicillin and streptomycin. The cells were plated on Matrigel/fibronectin-coated culture wells (100 μg/ml and 2 μg/ml, respectively; Sigma-Aldrich, St Louis, MI, USA) in the culture medium described previously 13 .
Transgene Excision in Culture
Immortalized cells in culture were transduced with pTrip CMV nlsCRE ΔU3 to excise immortalizing transgenes. The total amount of viral particles was 30 ng of p24 capsid protein per 105 cells. The cells were transduced in a minimum volume (300 µl) of RPMI 1640 culture medium containing 10% of decomplemented fetal bovine serum and 1% of penicillin and streptomycin. After 1 h of incubation at 37 °C with 5% carbon dioxide, the culture medium was added to complete the minimum volume (700 µl). Then, 24 h later, the medium was replaced by 1 ml of fresh medium. Photos of the cells were taken every week to evaluate the efficiency of excision by looking at cell morphology and cell growth. After 3 weeks, cells were plated on a LABTECH slide (Millipore, Burlington, MA, USA) to further evaluate the cell content by immunocytochemistry and quantitative PCR (qPCR) of RNA extracted from cell pellets.
ACT-164 Transplantation in Diabetic SCID Mice
Diabetes was induced in SCID mice by streptozotocin (Sigma-Aldrich) freshly prepared in citrate buffer and injected intraperitoneally to the mice in a dosage of 250 mg/kg body weight. Blood samples were collected from the tail at regular intervals over the next 2 days and used to measure glucose levels with glucose strips (ACCU-CHEK, Roche France, Boulogne-Billancourt, France) and a glucose meter. For the experiment, we selected mice whose blood glucose 1 to 2 days after the streptozotocin injection was above 3 G/l. To maximize survival, a LINBIT slow-release insulin capsule (LinShin, Scarborough, Ontario, Canada) designed for mice and lasting 2 to 4 weeks was implanted subcutaneously.
To evaluate the in vivo function of canine beta cells, 7 days after diabetes induction mice were transplanted with 2 million cells using an adapted published 11 procedure. Briefly, cells were placed in an Eppendorf tube and centrifuged for 3 min at 900×g. The medium supernatant was then removed and cells were resuspended in 10 µl of HBSS. The cell suspension was loaded in a polypropylene catheter (1 mm diameter) attached to a Hamilton pipet and next pushed into the center of the previously inserted O-ring.
In all, 22 mice were studied: eight served as controls (no graft), six received the immortalized ACT-164 beta cells, and eight received the same amount of ACT-164 cells after transgene excision. Blood glucose concentrations were measured at regular intervals in the morning after a 2-h fast. After 60 days, nephrectomy was performed in grafted mice to remove the transplants and assess their contribution to blood glucose control. The mice were sacrificed 24 h later. Each transplant was dissected from the adjacent kidney and studied by immunohistochemistry. In two mice from the transplanted and control groups, the pancreata were also dissected to assess beta-cell destruction by streptozotocin.
Immunofluorescent Staining
Grafted tissues were fixed in 3.7% formaldehyde, embedded in paraffin, and cut into 4-µm sections. Cells cultured on LABTECH slides (Millipore, Molsheim, France) were fixed in 4% paraformaldehyde, prepared in 1× PBS for 15 min at 4 °C and then washed twice in 1× PBS. Fixed samples were stained with guinea pig anti-insulin antibody (1/500; A0564-DakoCytomation, Carpinteria, CA, USA), rabbit antiglucagon antibody (1/300; 20076-Immuno, Euromedex, Souffelweyersheim, France), mouse anti-SV40LT antibody (1/50; DP02-Calbiochem, MerckBiosciences, Darmstadt, Germany), and mouse anti-human Ki67 antibody (1/50; DakoCytomation, Glostrup, Denmark). The secondary antibodies were fluorescein Texas Red anti-guinea pig antibody (1/500; 706-165-148-Interchim, Montluçon, France), antirabbit antibody (1/2000; A11070, Invitrogen, Carlsbad, CA, USA), and antimouse Alexa 488 antibody (1/1000; A11001, Invitrogen). Nuclei were stained with Hoechst 33342 fluorescent stain (1/20000; 62249, Thermo Fisher Scientific, Waltham, MA, USA). Digital images were taken using a Leica microscope or an Olympus FluoView FV1000 confocal microscope (Olympus, Shinjuko, Tokyo, Japan).
RNA Isolation and Quantitative Real-Time PCR (qRT-PCR) Procedure
Total RNA was isolated from the samples using the RNeasy Micro Kit 50 (Qiagen, Hilden, Germany; ref: 74004) according to the manufacturer’s instructions. First-strand cDNA was prepared using the Superscript First Strand Kit (Invitrogen, Carlsbad, CA, USA; ref: 11904-018). qRT-PCR was performed using LightCycler 480 SYBR Green I master mix (Roche Applied Science, ref: 04887352001) and analyzed on a LightCycler 480 Instrument II system (Roche Applied Science, Penzberg, Germany), according to the manufacturer’s instructions. The comparative method of relative quantification (2−ΔΔCT ) was applied to calculate the expression levels of each target gene, which were then normalized for dog glyceraldehyde 3-phosphate dehydrogenase mRNA. Primers used for qPCR have been described elsewhere 13 . Gene expression in ACT-164 cells was compared to that in adult dog islets using RNA extracts as reported elsewhere 16 .
Cell lysate for Insulin Content Measurement
To measure insulin content, cells were lysed directly in the culture wells with 20 mM Tris pH 8.0; 0.1% Triton X-100; 1% glycerol (TETG; 137 mM sodium chloride [NaCl]; 2 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid [EGTA]) and protease inhibitor tablet (Roche Applied Science) for 5 minutes on ice. The lysate was centrifuged at 3000 rpm for 5 min and stored at -20°C until insulin measurement. The cellular insulin content was measured in duplicate by ELISA.
Glucose-Stimulated Insulin Secretion
Insulin secretion was studied using nonexcised and excised ACT-164 cells cultured on 12-well plates. Four hours before starting the glucose stimulation, the cells were starved in HEPES-buffered Krebs-Ringer Buffer (KRB) (115 mmol/L NaCl, 5 mmol/L potassium chloride, 1 mmol/L calcium chloride, 1 mmol/L magnesium chloride, 24 mmol/L sodium bicarbonate, 10 mmol/L HEPES pH 7.4, and 0.2% bovine serum albumin) containing 2.8 mM of glucose. Stimulated insulin secretion was then measured during static incubation for 60 min in KRB containing first 2.8 mM then 15 mM of glucose. For the insulin content measurement, cells were lysed directly in the culture wells with TETG solution (20 mM Tris pH 8.0; 0.1% Triton X-100; 1% glycerol; 137 mM NaCl; 2 mM EGTA) and a protease inhibitor tablet (Roche Applied Science) for 5 min on ice. The lysate was centrifuged at 3000 rpm for 5 min and stored at −20 °C until insulin measurement. The insulin values in the supernatant were expressed as an absolute amount of insulin secreted for 60 min. Insulin secretion and intracellular content were measured in duplicate by ELISA.
Insulin Assay
Insulin was measured in duplicate in the media, cell extracts, and serum of SCID mice (see below) using a canine insulin ELISA (Mercodia, Uppsala, Sweden). The antibody used in this assay does not cross-react with mouse insulin. By using this ELISA kit, we were therefore able to detect and quantify dog insulin in serum from transplanted SCID mice.
Statistics
Statistical analyses were conducted using two-tailed Mann Whitney U-tests (GraphPad Prism 6) to compare insulin secretion by ACT-164 cells with 2.8 mM versus 15 mM glucose concentration. To compare blood glucose in mice with versus without ACT-164 cell transplants at all time points, a mixed-effect model compatible with repetitive measurements was used followed by post hoc Tukey’s multiple comparison test.
Results
Development of Fetal Canine Pancreas When Grafted in SCID Mice
In several animal species, as well as in humans, fragments of fetal pancreas grafted in SCID mice grow and develop. Whether this was the case for dogs was unknown and was therefore addressed in the first step of our research. SCID mice were grafted with several canine fetal pancreata obtained at 2-day to 3-day intervals from 41 to 55 days of gestation. The results clearly showed that at each stage studied, considerable development of the endocrine pancreas occurred in all the grafted specimens. Qualitatively, the endocrine development was very similar in all the specimens. In addition, the graft was already well developed 2 months after transplantation and continues to mature after 4 and 6 months. Figure 1 illustrates the results with three transplanted pancreata harvested after 41, 45, and 55 days of gestation, respectively. The grafted tissue was removed after 3.5 months (pancreas E45, top panel) or 5.5 months (middle and bottom panels). Insulin and glucagon endocrine cells were organized in aggregates of different sizes, although isolated cells were also observed. In some parts of the field, larger cell clusters resembling islets were identified.

Development of transplanted fetal pancreas E45 (top panel), E41 (middle panel), and E55 (bottom panel): fetal dog pancreata were transplanted in SCID mice, and the grafts were removed after either 3.5 or 5.5 months. Transplanted tissues were sectioned and then stained for insulin (red) and glucagon (green). The nuclei were stained in blue with Hoechst. Scale bars: 100 µm.
Production of Insulinoma and Expansion of Beta Cells
Canine pancreata dissected from 42-day-old to 55-day-old fetuses were first transduced with a lentiviral vector. Then, 2 to 6x fragments of each gland were transplanted under the kidney capsule of SCID mice. The grafts were removed 6 to 10 months later, and several cell lines were propagated. In all, pancreata of 36 litters were transduced, and more than 100 mice were grafted. A tumor was visible in only 30 mice. In all the other mice, the transplanted tissue was either absent or too small to be analyzed. We compared insulin and glucagon immunostaining in 6-month-old transplanted tissues originating from transduced or untransduced pancreatic fragments derived from 52-day-old fetuses from the same litter. As illustrated in Fig. 2, in untransduced tissue, the beta cells were numerous and intermingled with scarcer alpha cells (Fig. 2A–C). In contrast, in transduced tissue (Fig. 2D–F), the beta cells were found as very large aggregates, and only a few scattered alpha cells were visible (Fig. 2G–I). Furthermore, the SV40 large T antigen (SV40LT) was expressed in numerous cells, all of which also expressed insulin. The large number of cells positive for both SV40LT and insulin indicated the formation of an insulinoma. Several insulinomas that developed in four mice were used to derive cell lines after dissociation of the tumor, as described in the Materials and Methods section. We will now provide details on one of these lines, named ACT-164.

Expansion of insulinomas in transduced transplanted fetal pancreas The figure shows confocal images of stained 4-µm paraffin sections of transplanted pancreata from 55-day-old dog fetuses that were initially either not transduced (A–C) or transduced (D–I) with the lentiviral vector SV40LT under the control of the insulin promoter. (A–F) Sections were stained for insulin (red) and glucagon (green); the nuclei were stained in blue with Hoechst. (G–I) Sections were stained for insulin (red), SV40LT (green), and with Hoechst for the nuclei (blue): clusters of insulin-positive cells co-express the immortalizing transgene SV40LT. Scale bars: 100 µm.
ACT-164 Expresses Insulin and Proliferates
As shown in Fig 3, insulin was present in most ACT-164 cells. A few glucagon cells were visible and did not clearly co-localize with the insulin-positive cells. As expected, SV40LT was expressed in the nucleus of insulin-positive cells. As expected for an immortalized cell line, a large proportion of ACT-164 cells expressed the proliferation marker Ki67. The insulin content in ACT-164 cells was 0.55 ± 0.07 ng of insulin per million cells (n = 4).
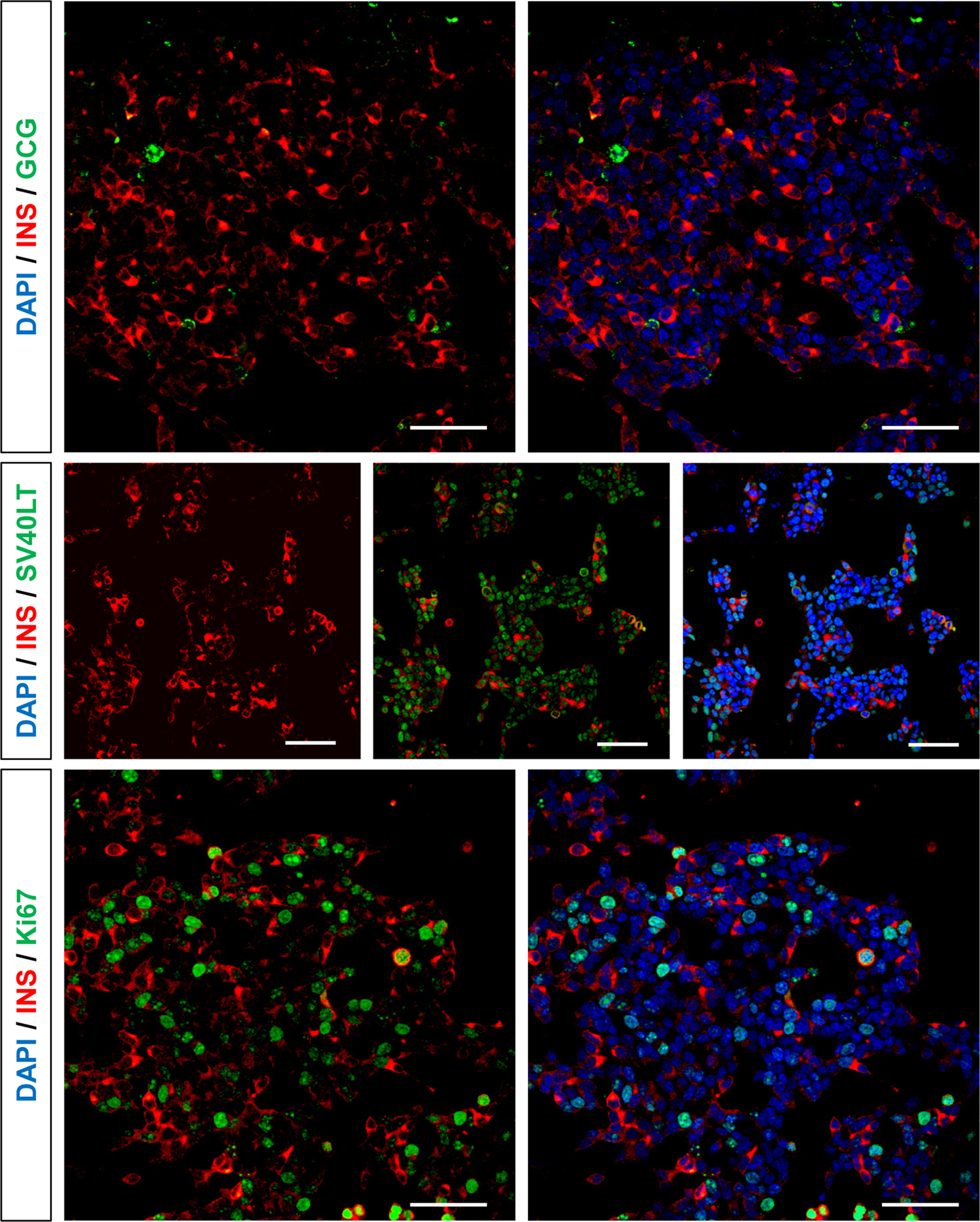
The ACT-164 cell line expresses insulin and SV40LT and proliferates ACT-164 cells in culture were fixed, stained, and imaged using a confocal microscope. Top panel: insulin (red) and glucagon (green); middle panel: insulin (red) and SV40LT (green); bottom panel: insulin (red) and Ki67 (green). All nuclei were stained with Hoescht (blue). Scale bars: 50 µm.
Cre-Mediated Excision of the SV40LT Immortalizing Transgene Resulted in Expansion Arrest
As described in the Materials and Methods section, the fetal pancreatic fragments were transduced with a lentiviral vector expressing an SV40 LT transgene that was excised upon Cre expression. ACT-164 cells were massively amplified, and the cells were then transduced with a Cre-expressing lentiviral vector. Untreated ACT-164 cells proliferated with a 5-day doubling time (Fig. 4, left panel). Following transduction with Cre, ACT-164 expansion was blocked (Fig. 4, left panel). In addition, Cre expression resulted in toxicity, and 3 weeks after transduction only 20% of the initial cells remained. Importantly, SV40 LT transgene excision had a profound morphological effect on the cells. Excised ACT-164 cells formed clusters, lost their neuron-like appearance, and acquired a round morphology (Fig. 4, middle and right panel).
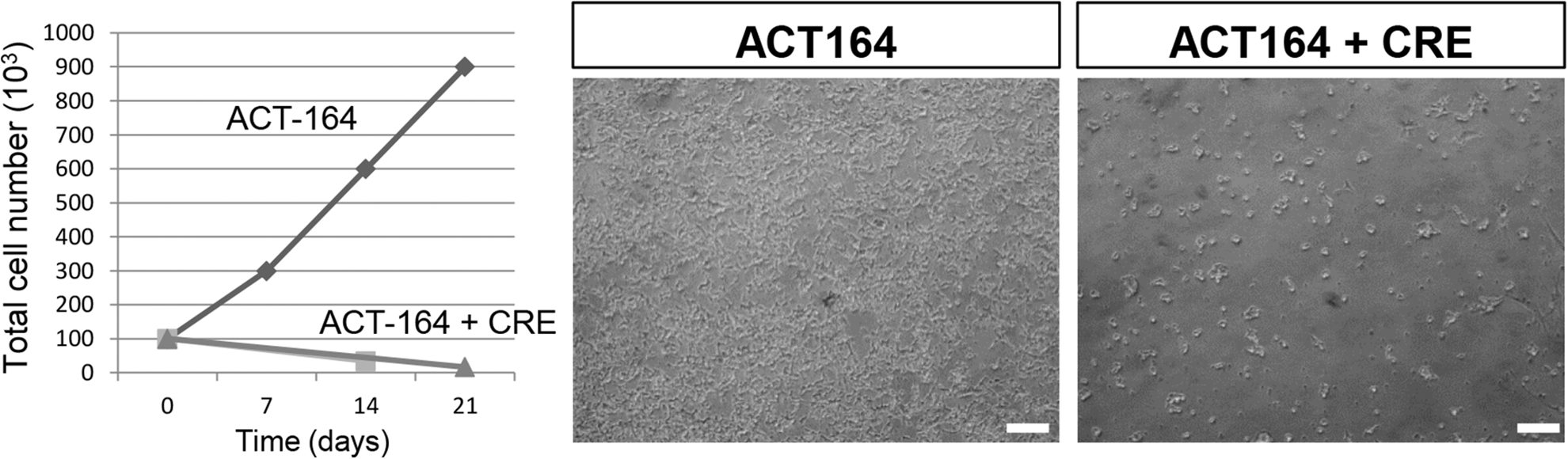
CRE transduction of ACT-164 blocks cell expansion and modifies cell morphology The total number of cells in culture was monitored for 21 days. ACT-164 cells expanded during this period, whereas ACT-164 cells treated with Cre on day 0 stopped their expansion and decreased in number over time. As illustrated in the Bright field images of cultured cells, cell morphology changed with Cre treatment from cells with a neuron-like shape dispersed in the dish to cells forming clusters. Scale bar: 100 µm.
Cre-Mediated Excision of ACT-164 Resulted in Enhanced Insulin Production
To address the effect of SV40 LT transgene excision, unexcised cells were compared with excised cells. Both types of cells were from the same batch, and the acquisition settings of the confocal microscope were strictly identical for both cell types. Figure 5 compares the immunostaining of unexcised (Fig. 5, left panel) and excised (Fig. 5, right panel) cells. Clearly, insulin staining was massively increased by excision. Importantly, the number of SV40LT-positive cells was markedly decreased, and the insulin staining was particularly intense in the SV40LT-negative cells. Furthermore, excised cells with marked insulin staining were negative for Ki67, indicating that they were unable to proliferate. The insulin content measured in extracts of four different batches of excised cells was 83.7 ± 14.7 ng insulin per million cells and was massively increased compared with the proliferating cells (152-fold increase).

Cre-mediated excision of immortalizing transgene blocks proliferation and enhances insulin expression. ACT-164 cells in culture treated or untreated (controls) with Cre were fixed, stained, and imaged using a confocal microscope. Settings were strictly identical across all samples for the red channel (laser intensity, PMT voltage, and gain). With such settings, insulin staining of the untreated ACT-164 cells was faint. The insert in the left top panel shows an image corresponding to the dotted area and taken with the microscope settings used in Fig. 3. With such settings, no insulin staining was visible in the untreated ACT-164 cells. Top panel: insulin (red) and glucagon (green); middle panel: insulin (red) and SV40LT (green); bottom panel: insulin (red) and Ki67 (green). All nuclei were stained with Hoescht (blue). Insulin staining was strongly enhanced by Cre treatment. Cells bearing an intense insulin signal were negative for SV40LT and Ki67 expression; some of them are indicated by a white arrow. Scale bars: 50 µm.
qRT-PCR in Excised or Unexcised ACT-164 Cells
To compare the relative expression of endocrine cell markers between excised versus unexcised ACT-164 cells, qRT-PCR was performed. With Cre treatment, ACT-164 cells expressed ninefold more insulin mRNA compared with untreated cells (Fig. 6A). This is consistent with the increase in insulin protein observed after Cre-mediated excision. In contrast, the expression of glucagon remained unchanged (Fig. 6B). In addition, the expression of the beta cell-specific transcription factor Pdx1 showed a significant 2.2-fold increase after Cre-mediated excision (Fig. 6C). This increase may partly explain the change in insulin expression. Although insulin expression increased sharply upon Cre-mediated excision, the insulin expression level was still 3500 times lower than observed in adult dog islets. The expression of Pdx1 mRNA in the control and excised ACT-164 cells was 10-fold and fourfold lower than in adult dog islets, respectively (Fig. 6C).

Comparative expression of insulin, glucagon, and Pdx1 transcripts upon excision of immortalizing transgene expression of insulin, glucagon, and PDX-1 was evaluated using quantitative real-time polymerase chain reaction in ACT-164 cells treated or untreated with CRE. The results were compared with the corresponding values found in pancreatic islets from an adult dog. Expression is normalized for glyceraldehyde 3-phosphate dehydrogenase. The results are reported as mean ± SEM from three independent mRNA preparations.
Insulin Production in Vitro and Effect of High Glucose Concentration on Insulin Secretion
Static glucose-stimulated insulin secretion experiments were performed in both control (proliferative) and excised cells (Fig. 7). Unexcised cells increased their insulin secretion when stimulated by 15 mM glucose (Fig. 7A), and the stimulation index (SI), defined as the ratio of secreted insulin at 15 mM glucose vs. at 2.8 mM glucose, was 2.7. When the same glucose stimulation was applied to excised cells, both basal and induced insulin secretions were increased 7.5-fold relative to the unexcised cells (Fig. 7B). The SI was 2.7 identical to that observed with unexcised cells.

Glucose-stimulated insulin secretion by ACT-164 cells before and after Cre-mediated removal of the immortalizing transgene. Static insulin secretion in response to glucose stimulation was measured using proliferating ACT-164 cells (A light grey) and ACT-164 cells treated with Cre 24 days before the measurement (B dark grey). In each culture well, cells were first incubated in KREBBS medium containing 2.8 mM glucose for 40 min and then 15 mM glucose for another 40 min. Results are expressed in ng of insulin secreted per hour and are mean ± SEM of four independent replicates. *Significant differences between 2.8 mM and 15 mM conditions (P value< 0.005).
Transplantation of ACT-164 Cells Normalized Blood Glucose in SCID Mice with Chemically Induced Diabetes
As shown in Fig. 8A, after the streptozotocin injection, blood glucose increased above 3 g/L in all animals. The insulin implants were highly effective, normalizing the blood glucose levels of all three groups of severely diabetic mice within a few days. In the SCID mice that did not receive cell implants, blood glucose increased gradually, exceeding 5 g/L after 2 months. By contrast, in the two groups implanted with ACT-164 cells, the blood glucose results were different. In the SCID mice implanted with unexcised ACT-164 cells, blood glucose remained almost within the normal range and differed significantly from that in the control group during the last 3 weeks of the experiment. In the group implanted with Cre-treated ACT-164 cells, blood glucose increased, reaching 3 g/L 5 weeks after implantation then decreased to values significantly lower than normal. After 2 months, four mice in this group had normal blood glucose concentrations. The implanted cells were removed by nephrectomy and studied by immunocytochemistry. As shown in Fig. 8B, these cells were positive for insulin but negative for glucagon. Unfortunately, all animals but one died within a few hours after nephrectomy, precluding a study of the effect of graft removal on blood glucose. The severity of beta-cell destruction by streptozotocin injection is illustrated in Fig. 8C. The pancreas of a grafted animal was studied after pancreatectomy. The tissue was almost devoid of beta cells and contained numerous glucagon-positive cells.
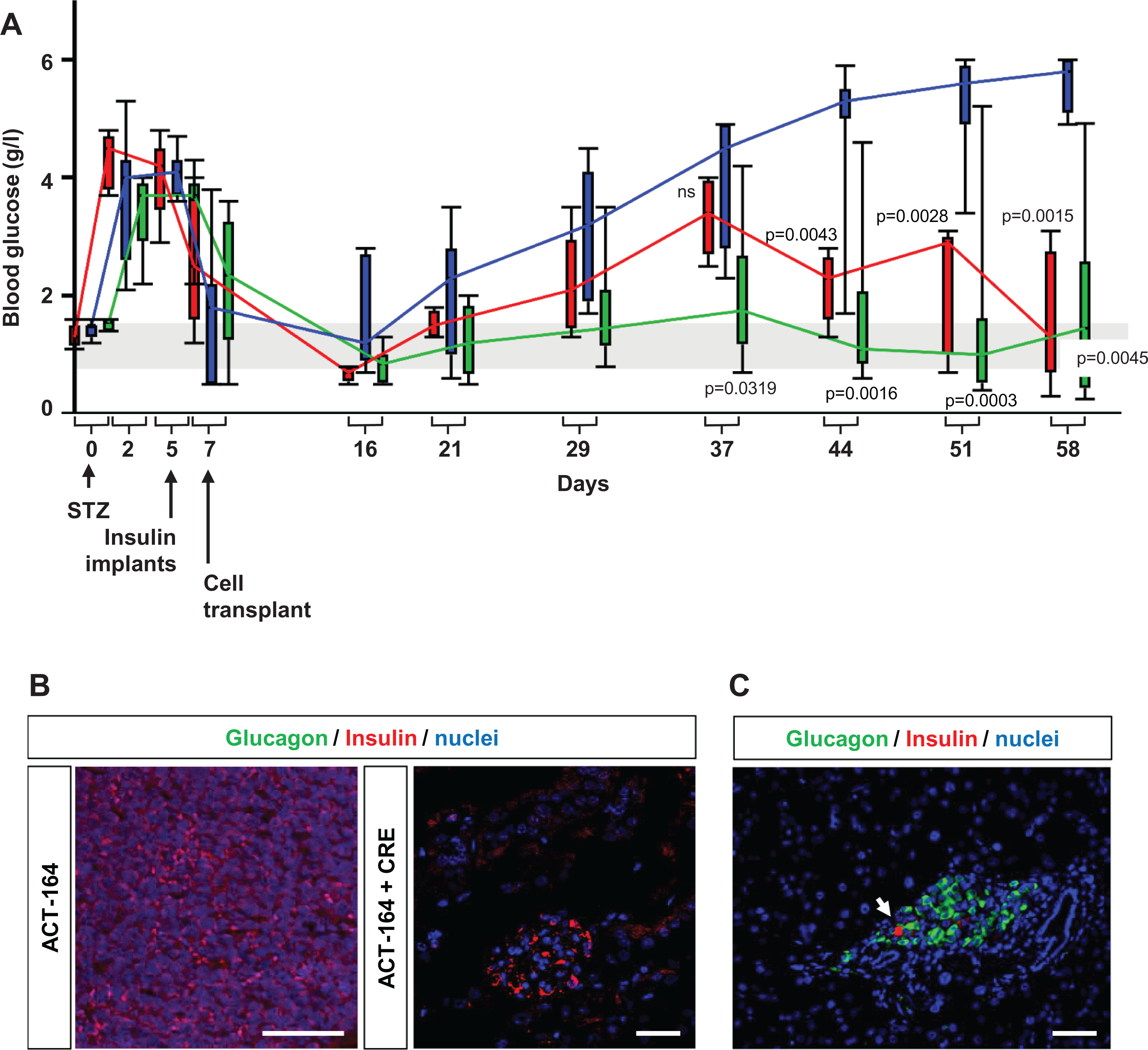
Transplantation of ACT-164 cells normalizes glycemia in diabetic SCID mice. (A) In 22 SCID mice, diabetes was induced by an intraperitoneal streptozotocin (STZ) injection. Implants releasing insulin for 2 to 4 weeks were inserted 5 days later. At the end of the first week, 2 million ACT-164 cells treated or untreated with Cre were transplanted under the kidney capsule of eight and six mice, respectively. The other eight mice served as controls. Blood glucose levels were measured at regular intervals in the three groups of mice: controls (blue), untreated ACT-164 cells (red), and ACT-164 cells treated with Cre (green). Box plots for each time points are represented. The whiskers show the range and the boxes represent the second and third quartile. For each group, the colored connecting line is going through the median of each time points. For statistical analyses, mixed-effect model compatible with repetitive measurements. Blood glucose were found to be significantly different with time (P < 0.0001 and F (4.694, 78.86) = 23.96) and with transplantation status (P < 0.0001 and F (2, 17) = 17.08). Interaction between time and transplantation status was also significantly different (P < 0.0001 and F (20, 168) = 8.106). We compared for each time point both transplanted groups with either ACT-164 cells or with excised ACT-164 cells with the untransplanted group using post hoc Tukey’s multiple comparisons test. Adjusted P values are indicated on the graph. The range of normal mice glycemia is indicated in grey. (B). Insulin (red) and glucagon (green) staining of the graft removed after 2 months. The transplanted tissue is clearly visible, with multiple insulin-positive and scarce glucagon-positive cells. (
Discussion
We have developed a canine beta-cell line using an approach adapted from an experimental design that was previously successful in generating a collection of immortalized rat 10 and human beta-cell lines exhibiting glucose-responsive insulin secretion 11,12 . Here, our approach was also based on targeted oncogenesis in fetal tissue, with the transduction of canine fetal pancreases with lentiviral vectors expressing a single immortalizing transgene, SV40 LT, under the control of the insulin promoter.
An important step in this procedure is the transplantation of the transduced fetal tissue under the kidney capsule of a SCID mouse, to allow full differentiation and maturation of the endocrine tissue and to generate an insulinoma due to the restricted expression of SV40LT in insulin-producing cells. Previous studies have shown that immune incompetent mice offer a permissive environment for the development of the fetal pancreata from humans 17 , rats 11,18 , sheep 19 , and pigs 20 . In this work, we were able to add the canine fetal pancreas to this list. When canine fetal pancreata of different gestational ages were grafted, without prior transduction, under the kidney capsule of SCID mice, the endocrine tissue developed and differentiated within a few weeks, resembling what can be observed in the endocrine pancreas of a 3-month-old pup 21 . The ability to replicate the differentiation of dog pancrease in vivo via transplantation in mice was the crucial first step in achieving the production of insulinomas from transduced canine fetal pancreatic tissue.
Pancreatic buds develop at the end of the first trimester of gestation in humans, whereas the canine fetal pancreata were collected during the last third of fetal development, after 42 days of gestation. We learned from a previous study 21 that the different steps of pancreatic development in dogs differ significantly from those in humans. The canine fetal endocrine pancreas is far less mature compared with its human counterpart. For example, at the end of gestation, the canine endocrine tissue contains chiefly alpha cells, with a few beta cells organized in clusters and no established islet structures. Although this has not been fully demonstrated in fetal dogs, the canine pancreas in the last third of gestation probably contains an important population of endocrine progenitors destined to differentiate and mature after birth. We therefore hypothesized that the oncogene would integrate into progenitor cells in the fetal pancreatic bud and that, being under the control of the insulin promoter, it would be expressed only by beta cells. A few months after transduction and transplantation, a large number of cells positive for both insulin and SV40LT were detected by immunolabeling, indicating that the process was efficient in allowing, in an ordered manner, transgene integration followed by SV40LT expression in beta cells and finally by the proliferation of beta cells as monitored with Ki67 staining. The resulting proliferative beta cells probably originated from transduced endocrine progenitors, although we cannot exclude that preexisting beta cells present at the moment of transduction were also targeted by the viral vector. Importantly, our previous results obtained with human tissue indicated that mature beta cells could not be efficiently immortalized by direct expression of SV40LT 11 . Insulinomas were obtained at all developmental stages from 42 to 55 days. However, some tumors were too small to serve as a source of cells for culturing. Also, cell line propagation failed with some tumors. In all, we obtained four insulinomas during more than 50 experiments with transduced fetal pancreata. Four cell lines were obtained, including ACT 164, which is characterized in this report.
Most ACT-164 cells were stained for insulin. A few cells expressed glucagon. Both the insulin mRNA level and the insulin content of this beta-cell line were considerably lower (by four orders of magnitude) than those measured by quantitative PCR in adult dog islets or extrapolated from cryopreserved adult canine islets 20 . Interestingly, the insulin content was also lower than the content previously reported in genetically engineered human beta cells 11,12 . Nevertheless, ACT-164 cell function mimicked that of a normal insulin-secreting cell, with insulin secretion being stimulated by an elevated concentration of glucose. When grafted to diabetic SCID mice, ACT-164 cells were able to normalize the blood glucose concentration within a few weeks. The cells expressed SV40LT and proliferated, whereas adult beta cells show very little proliferation. In the present work, to produce the ACT-164 cells, we engineered an integrative lentiviral vector that removed the immortalizing transgene upon Cre recombinase expression. The ACT-164 cells therefore constitute a conditionally immortalized canine beta-cell line. When the transgene was removed, the insulin content increased 150-fold and the corresponding mRNA expression level increased 10-fold. Importantly, we have observed that in both unexcised and excised cell populations, the insulin secretion ratio between 15 mM and 2.8 mM is identical, suggesting that both cell populations are able to respond to glucose. We also observed that the absolute amount of insulin secreted in basal and stimulated conditions is increased 7.5 fold in excised cells compared with unexcised cells. In addition, since the hallmark of excision is the absence of SV40LT expression, we have observed that SV40LT negative cells express a high level of insulin. Such cells do not exist in the control ACT164. Collectively, our results suggest that upon removal of SV40LT through CRE-mediated excision, the cells at the individual level express an increased level of insulin. At the population level, we observed that insulin content and expression is increased. Thus with an increased content but with a similar glucose responsiveness, the amount of secretion is increased with excision (basal and stimulated), and the insulin secretion index is similar in both conditions.
When transplanted in SCID mice with chemically induced diabetes, the ACT-164 cells rapidly normalized the blood glucose levels. Insulin secretion is difficult to compare between ACT-164 cells and normal canine islets, due to major protocol differences. Secretion data on adult dog islets were generated using a dynamic perfusion assay 22,23 , whereas, in this study, a static secretion assay was used. Nevertheless, the secretion index obtained with perfused cryopreserved adult dog islets was six, that is, twofold higher than that of our ACT-164 cells.
Working with dog tissue has some strong limitations. Most of the tools developed for islet studies were produced for rodent and human islets. Fortunately, antibodies developed for humans are appropriate for dogs, including those to insulin, glucagon, and Ki67. It would have been of interest to qualitatively evaluate the expression of Pdx1 and neurogenin 3, but neither of these dog compounds was recognized by human antibodies. We were able to quantitatively evaluate insulin, glucagon, and Pdx1 mRNA but not the other markers of interest. The dog neurogenin 3 sequence is incomplete in publicly available databases, and consequently, specific probes to detect dog neurogenin 3 could not be designed.
The clinical features of advanced diabetes in dogs resemble those observed in humans. Similar to human type I diabetes, the disease is due to a severe lack of insulin production reflecting nearly complete destruction of the pancreatic beta cells 3 . Whereas in humans, type 1 diabetes is currently viewed as an autoimmune disease, and several arguments indicate a different origin to the beta-cell destruction in dogs. In type 1 diabetes, insulitis is an important feature in humans but is lacking in dogs 3 . The circulating islet-cell autoantibodies commonly observed in humans with type 1 diabetes are usually not found in the serum of dogs at the time of diagnosis 24 . Although genetic studies have shown that variant alleles of the major histocompatibility complex are associated with the disease risk in dogs 25,26 , the autoimmune origin of canine IDDM is still under debate. Until the exact etiology of canine diabetes is clarified, it will be difficult to generate hypotheses about obstacles to graft survival. If beta-cell destruction is not due to autoimmunity in dogs, graft survival might depend only on natural immunity and, therefore, might be easier to achieve. In the absence of a better insight into canine IDDM etiology, it is difficult to speculate and predict the difficulties and success of beta-cell therapy in dogs. This is an important field to study and of great interest also for human type 1diabetes cell therapy. In conclusion, we have produced the first functional canine beta-cell line. We believe that this is an essential step that should advance diabetes care not only in animals but also in humans.
Footnotes
Acknowledgments
We thank Alice Pailleret and Noëmie Guedj for their excellent technical assistance in developing the cell culture model. This research work benefited from equipment and services from the Celis and iVector platforms at the Paris Brain Institute supported by Programme Investissements d’Avenir [ANR-10-IAIHU-06]. We are especially indebted to Océane RIBAULT and Laetitia STREHL for the ACT-164 culture and transduction experiments.
Ethical Approval
Ethical Approval is not applicable for this article.
Statement of Human and Animal Rights
All procedures in this study were conducted in Alfort Veterinary School (facilities 947-046-2), in accordance with the national review board CNREEA number 16, who approved the protocol (APAFIS n°2015042112442132).
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: PC and PR hold shares in Biotech “Animal Cell Therapy”.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research project was partly supported by funding from the BPI-France AIMA Program under the grant agreement #DA15005Q00/05, and from Biotech “Animal Cell Therapy”.