
Abstract
Hematopoietic stem cell (HSC) transplantation can be a potential cure for hematological malignancies and some nonhematologic diseases. Hematopoietic stem and progenitor cells (HSPCs) collected from peripheral blood after mobilization are the primary source to provide HSC transplantation. In most of the cases, mobilization by the cytokine granulocyte colony-stimulating factor with chemotherapy, and in some settings, with the CXC chemokine receptor type 4 antagonist plerixafor, can achieve high yield of hematopoietic progenitor cells (HPCs). However, adequate mobilization is not always successful in a significant portion of donors. Research is going on to find new agents or strategies to increase HSC mobilization. Here, we briefly review the history of HSC transplantation, current mobilization regimens, some of the novel agents that are under investigation for clinical practice, and our recent findings from animal studies regarding Notch and ligand interaction as potential targets for HSPC mobilization.
Introduction
E. Donnall Thomas was a pioneer in applying experience from animal bone marrow (BM) transplantation on leukemic patients. In 1959, he and his colleagues reported 3-month remission in a patient with total body irradiation followed by infusion of identical twin’s marrow 1 . Though Thomas’ finding was exciting, it was limited to isologous marrow infusion. In 1960, however, allogeneic transplantation was accomplished after human leukocyte antigen (HLA), the major histocompatibility complex, was discovered. An immunodeficient patient received marrow transplantation from an HLA-matched sibling without rejecting the allograft 2 . A decade later, after Thomas’ success, he and his colleagues cured some patients with end-stage acute leukemia by using a combination of chemotherapy, that is, cyclophosphamide (CY), with total body irradiation, followed by BM infusion from HLA-identical siblings 3 . Furthermore, the ability of donor lymphocyte to eliminate the tumor cells that have survived after ablative therapy was found to contribute to the reduction in the incidence of leukemic relapse after allograft transplantation. However, these lymphocytes may also contribute to a phenomenon called graft-versus-host disease 4 .
At the same time, other animal experiments discovered that nonleukemic blood cells egress from BM to peripheral blood system giving rise to mature blood cells and compensating damaged BM to maintain hematopoietic homeostasis 5,6 . These hematopoietic cells act as substitute sources to BM progenitor cells. However, Lewis and colleagues in 1968 suggested that due to the low frequency of the repopulating peripheral blood leukocytes (estimated to be 1/100th that of marrow), grafting would not be accomplished using these cells for transplantation 7 . Fortuitously, around the same time, a continuous flow centrifuge (apheresis) machine was developed to improve harvesting enough peripheral repopulating units for transplantation in patients with chronic myelocytic leukemia 8,9 . Nevertheless, several subsequent attempts to transfuse peripheral white blood cells among identical twins did not give a plausible engraftment result 8,10 . Putatively, the reason for the failure was due to the insufficient number of hematopoietic stem and progenitor cells (HSPCs) present in the peripheral blood compared to those in the BM.
Cryopreservation technique was developed in the 1970s and was found effective in preserving the hematopoietic stem cells (HSCs) from the peripheral blood 11 . Years later, using cryopreserved cells obtained from multiple rounds of apheresis before the transplantation resulted in successful hematopoietic engraftment 12 –16 . Despite many successes of cryopreserved HSPC infusion, harvesting a sufficient number of peripheral blood stem cells (PBSCs) for multiple rounds over a long time of collection was not feasible. However, the subsequent introduction of the course of CY and adriamycin chemotherapy resulted in ∼20-fold increase of PBSC yield, suggesting that chemotherapy could enhance PBSCs mobilization 17,18 .
In this review, we will first address the current HSPC-mobilizing agents, including chemotherapy, granulocyte colony-stimulating factor (G-CSF), and plerixafor, and other reagents that are still being investigated at various stages but not yet applied clinically. We will then discuss our recent animal research findings demonstrating the enhanced potential of HSPC egress and mobilization by blocking Notch or Notch ligands.
G-CSF and Chemotherapy
The clinically approved growth factor G-CSF has been widely used to mobilize both HSC and hematopoietic progenitor cell (HPC) for transplantation. G-CSF is a cytokine that promotes granulocyte proliferation and differentiation and enhances mobilization through direct or indirect protease activation. After G-CSF administration, neutrophil releases neutrophil elastase, cathepsin, and matrix metalloproteinase-9 (MMP-9), and the cell surface protease dipeptidyl peptidase 4 (DPP-4, cluster of differentiation (CD) 26) cleaves the adhesion molecules on HSPCs. These molecules, including stromal cell-derived factor 1 (SDF-1)/CXC chemokine receptor type 4 (CXCR4), stem cell factor (SCF; also called c-kit), and vascular cell adhesion molecule-1 (VCAM-1)/ very late antigen-4 (VLA-4), are responsible for HSCs retention in the BM 19 –23 . In addition, G-CSF indirectly inhibits osteoblast (OB) activity and reduces SDF-1 (also called CXCL12) expression that enhances HSPC mobilization 24 . Administration of G-CSF benefits both autologous and allogeneic mobilization, increases engraftment rate, and reduces hospitalization length and cost 25 –28 . The minimum target dose (≥2 × 106 CD34+ cells per kg weight) could be successfully achieved by 5–10 μg/kg of G-CSF per day for 5–7 days with one or more days of apheresis. However, a study led by Mayo Clinic demonstrated less than optimum mobilization in two groups of patients: non-Hodgkin lymphoma (71%) and multiple myeloma (30%) 29 . In autologous stem cell transplantation, chemotherapy is commonly used along with G-CSF to reduce the tumor burden, to enhance mobilization as well as to decrease the apheresis sessions and transfusion volumes. In myeloma patients, an intermediate dose of CY effectively mobilized HSPC 30 . On the other hand, in lymphoma patients, combination chemotherapies include G-CSF with CY, etoposide, dexamethasone, high-dose cytarabine, cisplatin (DHAP), and other chemotherapy regimens 31 –35 are often required for effective mobilization. Therefore, new agents with improved ability to increase stem cell yield and to reduce the number of apheresis sessions are required, particularly for those heavily pretreated patients.
Plerixafor
Plerixafor, or Mozobil (AMD3100), is the antagonist of CXCR4. AMD3100 prevents CXCR4 on HSCs from interacting with SDF-1 on BM stroma 36 –38 . BM OB, mesenchymal stem cells, and CXCL12-abundant reticular cells secrete SDF-1 to maintain HSC retention 39 , and disrupting this interaction between CXCR4 and SDF-1 leads to the mobilization of HSPCs to the periphery. AMD3100 administration not only blocks CXCR4, but also induces activation of MMP-9 and serine protease urokinase-type plasminogen activator 40,41 . Currently, plerixafor is clinically approved to use alone or in combination with G-CSF to mobilize HSPC in heavily pretreated lymphoma and myeloma patients 42 –47 . Although plerixafor is successful in increasing optimal CD34+ yield, decreasing mobilization failure, and reducing the number of apheresis sessions, its universal use is limited by its expensive cost 48 –53 .
Other Mobilizing Agents
Studies in mice have shown that two main axes are targeted directly or indirectly by most mobilizing agents, CXCR4 and the integrin VLA-4 signaling, or both 54 . Integrins are a family of proteins implicated in HSPC mobilization. Integrin adhesion receptors such as VLA-4 found on HSCs tether with VCAM-1 within the BM stroma 55 –57 . Indeed administration of anti-VLA-4 led to the mobilization of HSPC to the peripheral blood 58,59 .
While some agents, like G-CSF, target both pathways directly or indirectly, some only affect one adhesion molecule. An example of an approach that affects both CXCR4 and VLA-4 signaling is parathyroid hormone, which increases the secretion of G-CSF. In turn, this results in an increase in peripheral circulating HSC by 1.5- to 9.8-folds in mice 60 , and in a phase I clinical trial study, it induced adequate mobilization in 40%–47% of patients who have failed prior mobilization for autologous stem cell transplantation 61 . Sphingosine-1-phosphate (S1P), an agent mainly targeting the CXCR4 signaling, is a bioactive lipid mediator mainly found in red blood cells and acts as a ligand to G-protein-coupled S1P receptors. A high concentration of plasma S1P creates a gradient and egresses HSPC 62 –65 . Furthermore, during mobilization, HSPC egress is further mediated by the S1P gradient increase in peripheral blood as a result of erythrocyte lysis by the complement cascade and the membrane attack complex activation 66,67 . An alternative adhesion molecule target exists for proteasome inhibitors, such as bortezomib, which affects angiopoietin levels. Bortezomib successfully stimulated HSC mobilization in 85% of multiple myeloma patients receiving a bortezomib-based induction regimen 68 . Other agents or approaches that are currently undergoing investigation include cytokines, chemokines, chemokine receptor antagonists, bacterial toxins, proteases, inhibitors of adhesive cell interactions, ephrinA3 receptor antagonists, polymeric sugar molecules, prostaglandin inhibitors, blockade of heparan sulfate, Nlrp3 inflammasome activation, and stabilization of hypoxia-inducible factor-1α, among others 54 . Table 1 summarizes various agents that have been used in mice and their target adhesion molecules.
Mobilizing Agents (Not Including G-CSF and Plerixafor) and Their Target Adhesion Molecules.

CD: cluster of differentiation; CXCR: CXC chemokine receptor type; CXCL12: C-X-C motif chemokine 12; FLT3: fms like tyrosine kinase 3; G-CSF: granulocyte colony-stimulating factor; IL: interleukin; PGG-glucan: Poly-[1-6]--D-glucopyranosyl-[1-3]--D-glucopyranose glucan; SDF-1: stromal cell-derived factor; VCAM-1: vascular cell adhesion molecule; VEGFR2: vascular endothelial growth factor receptor 2; VLA: very late antigen.
Systemic Factors
Several factors affect HPSC mobilization on the systemic level. These include cortisol level, the time of day, stress, exercise, trauma, infection and inflammation, elements in coagulation and complement cascades, and signals from the sympathetic and central nervous systems 54 . These factors may contribute to the overall stem cell collection during apheresis procedures.
Notch2 and Notch Ligand Blockade
Notch signaling plays a critical role in multiple pathways that control cell fate determination, such as embryogenesis, neurogenesis 117 –120 , angiogenesis 121 , cardiogenesis 122 , and hematopoiesis 123 . In hematopoiesis, this signaling not only regulates lymphopoiesis but also regulates myelopoiesis 124,125 . Notch pathway alterations play critical roles in several cancer types and particularly in hematologic malignancies. Therefore, it is a desirable target in these neoplasms. However, inhibiting a downstream enzyme, the gamma-secretase, in this pathway, has not been successful due to side effects from global inhibition of wild-type Notch proteins. On the other hand, targeting specific Notch proteins, for example, Notch1 or Notch2, may still be promising 126 . An essential feature of Notch is its adhesive nature, which was first described by cell aggregation assays in Drosophila 127,128 . However, the precise role and the biological significance of Notch receptors and ligands as adhesion and signaling molecules in HSC biology, particularly in the context of HSC cell therapy, have not been well defined. Here we will report our recent findings suggesting that Notch2 and its interaction with Notch ligand may serve as potential effective HSPC-mobilizing targets.
In mammals, the canonical Notch signaling pathway is initiated by binding interactions between the extracellular domain of a Notch family member (Notch1–4) on a receiving cell and a Notch ligand of the Jagged (Jagged 1 and 2) or Delta-like (DLL1, 3, and 4) families on a sending cell 129 . It is generally accepted that ligand binding initiates ligand endocytosis and successive proteolytic cleavages, which culminate in the release of the intracellular domain of Notch and the formation of a large transcriptional activation complex leading to the activation of downstream targets of Notch signaling 130,131 . Despite in vitro evidence that activation of Notch stimulates HSC self-renewal 132 –135 , the in vivo function of Notch in HSC is still debatable. Conditional deletion of Notch receptors, ligands, or Notch canonical targets does not appear to affect HSC steady-state homeostasis 136,137 . In contrast, Notch2 was found responsible for the rate of generation of repopulating stem cells during stress hematopoiesis and the early phase of hematopoietic recovery 138 . Also, Jagged1 expressed by BM endothelial cells regulates homeostasis and regenerative hematopoiesis, while DLL4 expressed by osteocalcin-expressing bone cells is responsible for generating early thymus progenitors 139,140 .
We and others found that Notch2 is the primary Notch receptor expressed on HSCs and nonlymphoid committed progenitors. In comparison, Notch1 expression level is low on HSC cells but high on lymphoid progenitors 141,142 . Notch transactivation is the result of the engagement of Notch receptors with Notch ligands. This process is dependent on posttranslational modification of Notch receptors with O-glucose and O-fucose, added to serine in the consensus C1-X-S-X-(P/A)-C2 or to serine or threonine residue in the consensus sequence C2-X-X-X-X-(S/T)-C3 143 –145 , by protein O-glucosyltransferase 1 (Poglut) or protein O-fucosyltransferase 1 (Pofut1), respectively, on the epidermal growth factor (EGF) modules of Notch extracellular domain 146 –149 . O-Fucose, but not O-glucose, enhances Notch affinity for Jagged or Delta-like ligands and regulates Notch signaling transactivation 147,150 –152 . This notion is supported by crystal structural determinations showing that O-fucose attached to the Thr residue of Notch1 EGF-like repeat acts as a surrogate amino acid to make functional contact with a specific domain on DLL4 (contact Notch1 EGF12) and Jagged1 (contact Notch1 EGF8 and 12), and thus directly affects Notch ligand binding 153,154 . Elongation of O-fucose to a disaccharide (GlcNAc-fucose) on EGF12 by any of three Fringe enzymes (N-acetyl-glucosaminyltransferase)-Lunic fringe (Lfng), Manic fringe (Mfng), or Radical fringe (Rfng) further increases Notch1 binding affinity to Jagged1 and DLL1 and affects Notch activity in slightly different ways 145,155 –158 .
To understand the role of Notch O-fucose glycan modification in its adhesive interaction with Notch ligand in the marrow HSC compartment, we studied mice with Pofut1 deletion, and thus O-fucose deficiency 125,141,159 . These mice developed increased HSC cycling and increased HSPC egress from the marrow manifested as neutrophilia along with an increase in circulating myeloid progenitors. The altered homeostasis of O-fucose-deficient HSC is accompanied by the more distal locations of Lin–c-kit+Sca-1+ (LSK) cells relative to the endosteum and the OBs when compared to control HSCs 159 . The increased cell-autonomous HSC cycling and egress are primarily accounted for by a loss of binding of O-fucose-deficient HSC to Notch ligands 141,160 . The loss of binding results in a decreased adhesion of Pofut1-deficient HSCs to marrow stromal cells. In the in vitro cell adhesion assay, wild type (WT) long-term HSCs (LT-HSC; CD48–CD150+LSK), but not O-fucose-deficient LT-HSCs from Pofut1-null mice, showed 15%–25% increased adhesion to a stromal cell line from mouse bone marrow (OP9) cells expressing Notch ligand (Jagged1, DLL1, or DLL4) relative to parental OP9 cells 160 . The recombinant ligand entirely blocked the Notch ligand-mediated adhesion. Further, co-culture with OP9-DLL1, OP9-DLL4, or primary calvarium OBs increased the quiescent cell fraction of WT LSKs in G0 phase (from basal level 17% to 37%, 48%, and 67%, respectively), whereas Pofut1-null LSKs remained less quiescent on OP9-DLL1/DLL4 or primary calvarium OBs.
To examine the specific contribution of different Notch ligands and Notch receptors that support HSC quiescence and niche retention, we applied neutralizing antibodies targeting Notch ligand Jagged1 or DLL4. These antibodies block specific interaction of each ligand to Notch receptors 161,162 . Both Jagged1 and DLL4 are expressed in BM endothelial cells and OBs/osteolineage cells 133,140,163 –165 . In vivo, we found that circulating LSK and LK cells in the periphery of mice receiving anti-Jagged1 or anti-DLL4 increased 2.5- to 3.3-fold, respectively, compared to those receiving isotype control antibodies. White blood cells increased modestly, while platelet numbers did not change significantly in mice receiving anti-Jagged1 or anti-DLL4. There was an increase in circulating granulocytes and a decrease in T lymphocytes in mice receiving anti-DLL4 but not in mice receiving anti-Jagged1 160 . HSPC frequencies did not change, except that common lymphoid progenitors (CLPs) decreased in anti-DLL4-treated mice, consistent with the role of DLL4 in promoting CLP development in other reports 140 . We found that there was an increase in marrow HSPC proliferation following DLL4 but not after Jagged1 blockade. Further, mice receiving Jagged1- or DLL4-antibody followed by G-CSF (4 doses) and plerixafor treatment showed a further ∼50% increase in LSK mobilization relative to control-treated mice 160 .
More recently, we examined the effects of Notch receptor blockade. Unlike ligand neutralizing antibodies, Notch receptor-specific blocking antibodies do not interfere with receptor–ligand interaction, but instead block cleavage of Notch receptors and thus downstream signaling activation 161 . We found that in mice receiving Notch2-blocking antibodies, but not Notch1-blocking antibodies, spleen-residing LSKs and LKs increased three- to four-fold. When mice were given G-CSF and plerixafor following four doses of anti-Notch2 treatment, a 2.5-fold increase of white blood cells, a 3- and 3.3-fold increase of LSKs and LKs were seen in the periphery, and a 3.6- and 2-fold increase of spleen-residing LSKs and LKs were found in mice receiving anti-Notch2 compared to control-treated mice 116 . However, Notch2 blockade, combined with G-CSF or plerixafor, did not affect marrow HSPC homeostasis. We confirmed that increased HSPC egress following Notch2 blockade is a result of Notch2 signaling loss in the hematopoietic system, since Notch2 deletion in hematopoietic tissues caused increased cell-autonomous egress of HSPC to the PB and the spleen by reconstituted Notch2-deficient BM cells from Vav-Cre/Notch2F/F mice in wild-type recipients. However, the HSPC homeostasis was not much affected in the marrow of Notch2-deficient mice.
Similar to the effect of anti-DLL4, the quiescent G0 HSCs were decreased in mice receiving Notch2 antibodies or in the marrow of Notch2-deficient mice 116 . We show that transient Notch2 blockade or Notch2 loss in mice leads to decreased HSPC CXCR4 expression but increased cell cycling with CXCR4 transcription being directly regulated by the Notch transcriptional protein RBPJ. Surprisingly, we found that Notch2 blockade enhances HSPC homing and hematopoietic recovery. We observed that a 1.9-fold more of Notch2-blocked progenitors homed to the BM compared to their corresponding controls. We studied the hematopoietic reconstitution at various time points after the transplantation of cells from mice that received either control antibody, Notch1 or Notch2 blocking antibodies. Peripheral blood analysis revealed a better recovery of platelets at 4 and 8 weeks and a higher hemoglobin level until 10 weeks in recipient mice receiving Notch2-blocked cells than in mice receiving control-treated cells or Notch1-blocked cells. Analysis of BM 3 months after transplantation showed that the megakaryoerythroid progenitors and the common myeloid progenitors, derived from Notch2- but not Notch1-blocked donors, increased by ∼92% and 75%. At the same time, alteration to the HSC homeostasis did not occur.
In summary, either blocking Notch ligand adhesion through neutralizing Jagged1 or DLL4 or blocking Notch2 signaling induces HSPC egress and mobilization without significantly affecting HSC homeostasis (Fig. 1). In addition, transient Notch2 blockade results in higher engraftment and better myeloid reconstitution. The underlying mechanism is being investigated.
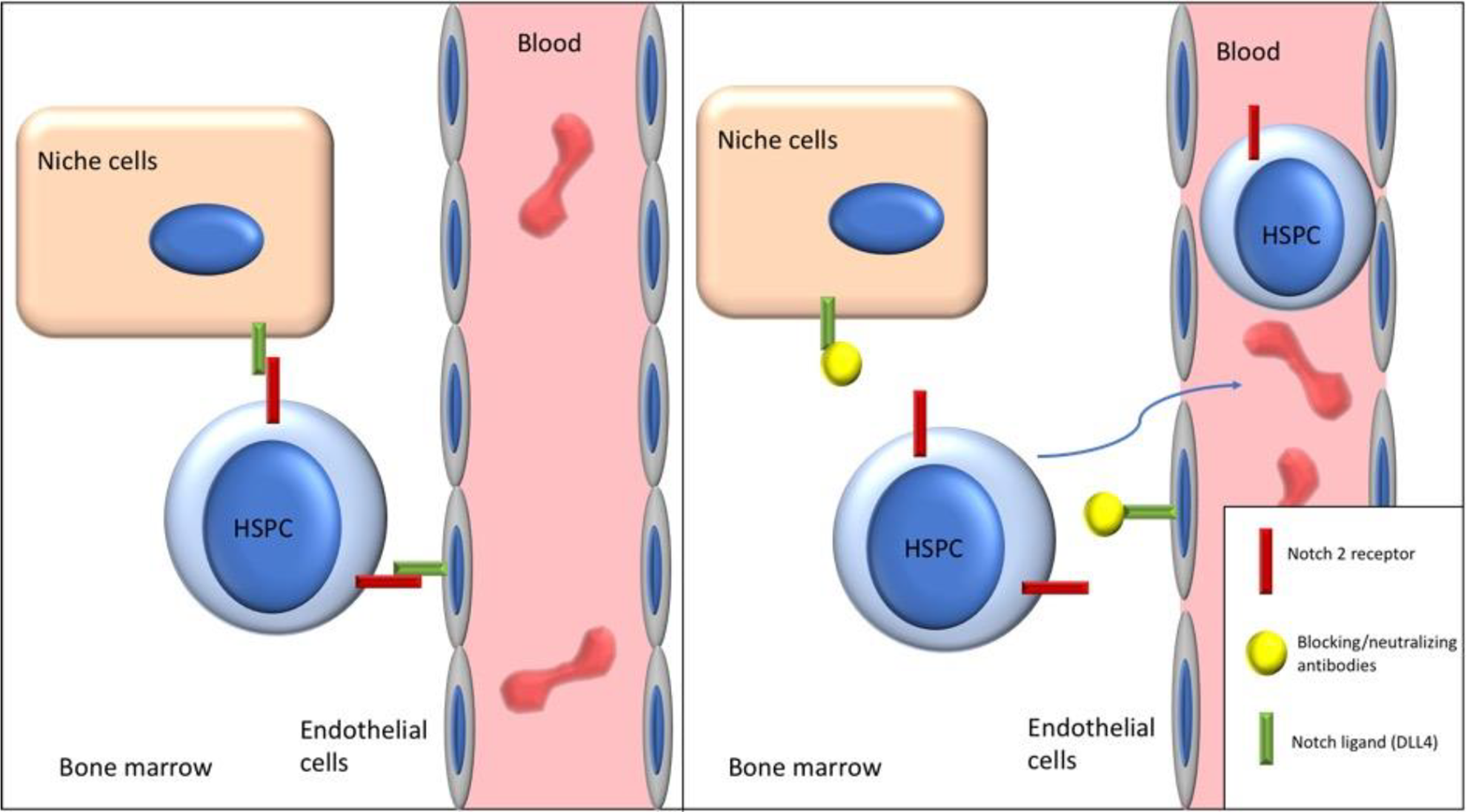
Blocking of Notch2-ligand adhesion through neutralizing and blocking antibodies induces HSPC egress and mobilization. DLL4: Delta-like 4; HSPC: hematopoietic stem and progenitor cell.
Conclusion
Hematopoietic cell transplant (HCT) is a potentially curative therapy for blood and nonhematologic disorders. A successful outcome is dependent on the infusion of an adequate number of functionally active mobilized HSPCs. Until recently, G-CSF, either alone or in combination with chemotherapy, failed to mobilize an optimal CD34+ cell dose (5 × 106/kg) in up to 40% of patients 166 . Plerixafor, in combination with G-CSF, increased total CD34+ cells mobilized and often is used for HPC mobilization in myeloma and non-Hodgkin lymphoma patients 167 . Unfortunately, this approach does not always lead to adequate HSPC collection in patients with prior extensive cytotoxic therapy. In some instances, multiple HCT procedures may require higher numbers of HSPCs 168 –170 . Newer mobilizing regimens, either alone or in combination with G-CSF and plerixafor, have shown improved collection efficacy in preclinical models and could facilitate the collection of sufficient cells for multiple transplants and significantly decrease procedure-related risks, such as thrombocytopenia and infection associated with large volume or multiple collections 171 –173 .
Footnotes
Author Contributions
Conceptualization: MA, LZ; literature search and data analysis: MA, HT, LZ; writing—original draft: MA, LZ; writing—review and editing: HT, LZ.
Ethical Approval
Our institution does not require ethical approval for reporting review articles.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by research funding from NCI CA222064 and NIH HL103827 (to LZ), and by the Department of Pathology Case Western Reserve University faculty startup fund to LZ.